

Design of the study
This study was a randomized, double-blind, placebo-controlled clinical trial (NCT: 06539260, 06/08/2024) that administered a three-month supply of vitamin D3 and a placebo(vegetable oil) to patients with PD who had low levels of vitamin D. This study obtained approval from the Ethics Committee of Suzhou Hospital of Anhui Medical University, with approval number A2023024. The procedures used in this study adhered to the principles of the Declaration of Helsinki. All patients signed informed consent to participate in the study and were informed of their right to withdraw at any point during the trial.
Participants
From January 2023 to March 2024, 51 PD patients were recruited. All subjects were evaluated and treated in the Parkinson’s Out-patient and In-patient department of the Neurology Department at Suzhou Hospital of Anhui Medical University. Fifty partners or volunteers of PD patients, matched by sex and age, are selected from Suzhou Hospital of Anhui Medical University to form a healthy control(HC) group. The inclusion criteria for patients were as follows: (1) Meet the diagnostic criteria for PD established by the UK Parkinson’s Disease Society Brain Bank. (2) Age between 45 and 80 years. (3) Have a disease duration of less than ten years. The exclusion criteria were as follows: (1) Have related vitamin D metabolic diseases (such as renal failure, severe liver damage, inherited 1α-hydroxylase deficiency, etc.). (2) Have immune system diseases. (3) Have a history of disabling cerebrovascular disease. (4) Have first- or second-degree relatives with PD. (5) Have severe dementia, depression, or severe mental illness.
Randomization and masking
For the PD patients included in the study, analyses were conducted on their peripheral blood vitamin D levels. PD patients with low vitamin D levels were divided into two groups at a 1:1 ratio to receive vitamin D3 and PL. Randomization was performed via an Excel random number generator. Both vitamin D3 and the PL were placed in identical bottles labeled only with numbers and containing no additional information. The patient is administered either one 5000-unit Vitamin D2 softgel capsule or one placebo capsule orally every 5 days. Product allocation and randomization were carried out by an independent researcher. All parties involved—including assessing physicians performing scale evaluations, data analysis personnel, and patients—remained blinded to treatment allocation until database lock. Unblinding occurred only after completion of the final statistical analysis.
Outcome
The primary endpoint of this study focused on the changes observed from baseline (T0) to the 3-month mark (T1). We evaluated PD patients via a comprehensive set of scales, including the Movement Disorder Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), Berg Balance Scale (Breg), Montreal Cognitive Assessment (MoCA), Mini-Mental State Examination (MMSE), Self-rating Anxiety Scale (SAS), Self-rating Depression Scale (SDS), and Parkinson’s Disease Sleep Scale (PDSS). All patients underwent scale assessments at 1.5 h after medication administration (the peak drug concentration period) in order to minimize the impact of the wearing-off phenomenon on the evaluation.
During the initial T0 stage, we analyzed the peripheral blood levels of Th17, Treg, and Vit D in the PD patients and healthy volunteers. Among the PD patients with low vitamin D levels, we randomly assigned them into two groups: one to receive vitamin D3 supplementation and the other to receive PL. At the T1 stage, we followed up with both groups of PD patients to reassess the indicators and monitor any changes (Fig. 1).
Fig. 1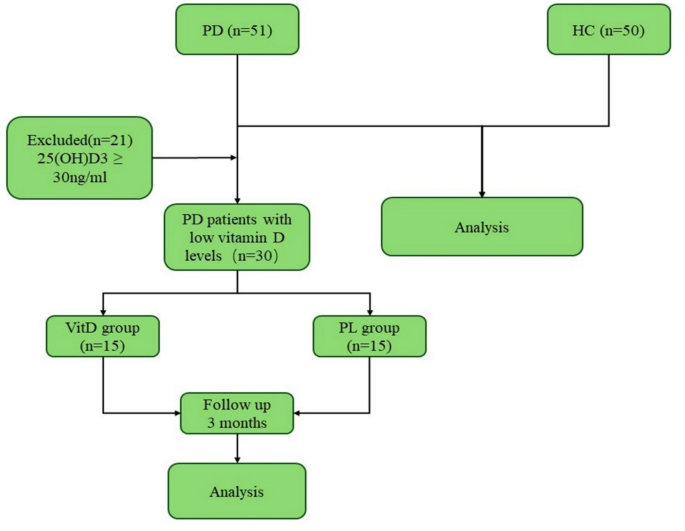
Flow diagram of the study.
Elecsys method
The serum 25(OH)D3 concentration was measured via the Elecsys method on a Roche electrochemiluminescence fully automated immunoassay analyzer. According to the guidelines of the Endocrine Society, normal vitamin D levels are defined as ≥ 30 ng/mL, 20–30 ng/mL is defined as insufficient, and vitamin deficiency is defined as ≤ 20 ng/mL25.
Flow cytometry
Venous blood withdrawal was performed after a night of fasting, between 8:00 a.m. and 10:00 a.m., into heparin lithium anticoagulant tubes. The tubes were subsequently coded and stored at room temperature until processing, which occurred 12 h after collection, to ensure homogeneous treatment of all the samples.
Blood was diluted 1:1 with RPMI 1640, mixed with leukocyte activation cocktail (3 µL/mL, BD Pharmingen, 550583), and incubated for 4–6 h at 37 °C under 5% CO2. After hemolysis (BD Pharmingen), cells were washed, surface-stained with FITC-conjugated anti-CD3 (BD Pharmingen, 555332) and PerCP-Cy5.5-conjugated anti-CD8 (BioLegend, 341051), fixed/permeabilized (BD Fixation/Permeabilization Kit, 554714), and intracellularly stained with PE-conjugated anti-IL-17 A (BD Pharmingen, 560436).
For Tregs, blood was surface-stained with CD4-FITC (BD Pharmingen, 566911) and CD25-PE (BD Pharmingen, 555432), lysed, fixed/permeabilized (eBioscience FOXP3/TRANSCRIPTION FACTOR STAIN BUFFER, 00-5523-00), and intranuclearly stained with Foxp3-APC (eBioscience, 17-4776-41).
Acquisition was then performed on a BD FACSCanto II flow cytometer. CD3+CD8−IL-17+ cells were identified as Th17 cells, whereas CD4+CD25+Foxp3+ cells were identified as Treg cells. Lymphocytes are recognized on the basis of their classical forward scatter (FSC) and side scatter (SSC) signals. The data were analyzed via FlowJo software (version 10.8.1).
Statistical analysis
In this study, SPSS 26.0 and GraphPad Prism v.9 software were used for data analysis. Descriptive statistical methods were employed to analyze all the data gathered during the research process. For intergroup comparisons, an independent sample t test was conducted, a paired sample t test was used for comparisons before and after the follow-up. Pearson correlation analysis was applied to data adhering to a normal distribution, whereas Spearman correlation analysis was employed for data deviating from a normal distribution. A p value of less than 0.05 was considered statistically significant.





