

Abstract
Glioblastoma (GBM) possesses a profoundly immunosuppressive tumor microenvironment (TME) dominated by innate immune mechanisms. Tumor-associated macrophages (TAMs), microglia, and myeloid-derived suppressor cells (MDSCs) constitute the major immunosuppressive axis, promoting tumor progression through cytokine secretion (IL-10, TGF-β), metabolic reprogramming, and inhibition of cytotoxic immunity. These innate immune cells not only facilitate immune evasion but also impair adaptive T-cell responses, limiting the efficacy of current immunotherapies. Emerging evidence highlights the therapeutic potential of targeting innate immunity via TAM repolarization, MDSC depletion, and NK cell activation to reshape the immunosuppressive TME. This review summarizes the pivotal role of innate immunity in GBM pathogenesis and explores novel combinatorial strategies that integrate innate immune modulation with checkpoint blockade, oncolytic virotherapy, and metabolic interventions to overcome therapeutic resistance in this lethal malignancy.
1 Introduction
Glioblastoma, the most common primary central nervous system (CNS) malignancy, accounts for 80% of primary malignant brain tumors (1). Although molecular advances have enhanced our understanding of GBM pathogenesis and led to improved treatment strategies, substantial gaps remain in translating these discoveries into effective clinical interventions (2–4). Despite advancements in surgical resection, radiation therapy optimization, and molecularly targeted treatments, clinical outcomes remain suboptimal. Consequently, there is a pressing demand for innovative therapeutic modalities and novel pharmacological interventions.
Cancer immunotherapy, particularly immune checkpoint inhibition, has revolutionized oncology by facilitating durable antitumor immune responses (5). Clinically, inhibitors targeting PD-1/PD-L1 and CTLA-4 have demonstrated efficacy in malignancies such as melanoma, non-small cell lung carcinoma, and renal cell cancer. However, their effectiveness in GBM remains constrained due to the unique immunosuppressive tumor microenvironment and sophisticated immune escape mechanisms (6–8). These findings indicate that monotherapy failure results from multifactorial mechanisms, including the accumulation of immunosuppressive cell subsets, dysregulated cytokine signaling, and impaired antigen presentation within the GBM microenvironment (9–11). Moreover, GBM cells utilize multifaceted immune evasion strategies, such as overexpressing immunosuppressive ligands and recruiting Tregs and MDSCs, which collectively undermine therapeutic efficacy (12). Consequently, comprehensive characterization of the glioma immune landscape and intricate cellular crosstalk within the tumor microenvironment is essential for developing optimized, patient-specific immunotherapeutic strategies.
2 Glioblastoma microenvironment2.1 Innate immunity2.1.1 Tumor-associated macrophages in glioma
Within the glioma immune landscape, TAMs represent the predominant immune population, consisting of both bone marrow-derived macrophages (BMDMs) and resident microglial cells (13, 14). Microglia arise from primitive yolk sac precursors and maintain their population through CSF1R-dependent self-renewal, whereas BMDMs are recruited from peripheral circulation primarily via the CCL2/CCR2 chemotactic axis (15). Historically, these subsets were distinguished by surface markers—CD11b+CD45^high for macrophages versus CD11b+CD45^low for microglia (16)—though recent single-cell transcriptomic studies have identified more refined signatures, including CCR2, CD45RA, and CD209 for macrophages, and CX3CR1, P2RY12, and TMEM119 for microglia (17). Morphologically, microglia display an extended, ramified structure, while macrophages appear more compact and highly motile (18). Although microglia serve as the primary immune sentinels in the CNS, BMDMs constitute the majority of TAMs in GBM, with microglia predominating in primary tumors and macrophages increasing in recurrent disease (16, 19). Elevated TAM infiltration is strongly associated with worse clinical outcomes. The functional plasticity of TAMs is a key focus in neuroimmunology (20).
Conventionally, these cells are categorized into M1 and M2 phenotypes. M1 macrophages are pro-inflammatory, induced by interferon-gamma and tumor necrosis factor-alpha, characterized by the expression of CD80 and CD86, and secrete reactive oxygen species, interleukin-1β, and interleukin-12. In contrast, M2 macrophages exhibit immunosuppressive properties, are polarized by interleukin-4 and interleukin-13, express CD206 and arginase-1, and produce interleukin-10, transforming growth factor-beta, and CCL17 (21, 22). However, high-resolution single-cell analyses have revealed considerable transcriptional overlap, which challenges the traditional binary classification and instead supporting a continuum of activation states (23). TAMs facilitate immune escape through multiple mechanisms, including MET–STAT4–PD-L1 signaling (24) and TLX-induced PD-L1 upregulation, which suppresses tumor-infiltrating lymphocytes (TILs) (25). Additionally, glioma-derived exosomes promote lipid accumulation in macrophages via TMEM198B, reinforcing M2-like polarization (26). Given the critical role of TAM heterogeneity in disease progression, personalized immunotherapeutic approaches targeting immunosuppressive TAM subsets may enhance treatment responses in glioma patients.
2.1.2 Neutrophils and NK cells in the glioblastoma microenvironment
As critical effectors of innate immunity, neutrophils represent nearly 70% of peripheral leukocytes and play context-dependent roles in glioblastoma pathogenesis (27). During initial tumor development, these cells exert antitumor effects by secreting antimicrobial peptides and pro-inflammatory cytokines, thereby enhancing immune-mediated tumor suppression. However, in advanced disease stages, the TME reprograms neutrophil activity, shifting their function toward promoting tumor expansion and metastatic dissemination (28). Within the TME, neutrophils engage in extensive crosstalk with other immune populations, secreting a variety of soluble factors that facilitate neoplastic growth, vascular remodeling, and the establishment of an immunosuppressive niche (29). NK cells, another vital component of innate immunity, are widely present in the GBM microenvironment, though their tumoricidal capacity is often severely attenuated (30). Malignant cells employ numerous immune escape mechanisms, including reduced expression of activating ligands and increased presentation of inhibitory signals, which collectively impair NK cell recognition and cytotoxic function (31). Additionally, immunosuppressive cytokines such as TGF-β and IL-10 within the TME further inhibit NK cell activity, reinforcing immune evasion. Nevertheless, NK cells retain significant therapeutic potential, as they can selectively target and eliminate treatment-resistant glioma stem cells while also enhancing antitumor immunity through mechanisms like antibody-dependent cellular cytotoxicity (ADCC) (32–34).
2.2 Myeloid-derived suppressor cells and dendritic cells
MDSCs constitute a diverse group of immature myeloid lineage cells exhibiting strong immunosuppressive properties. Increased MDSC populations have been observed in the circulation and tumor microenvironments of individuals with glioblastoma (35). These cells promote oncogenesis through multiple mechanisms, including the release of immunomodulatory cytokines and growth factors, inhibition of antitumor immune effector functions, and stimulation of vascular proliferation. Notably, IL-6 production by MDSCs is mediated through STAT3 pathway activation, which supports neoplastic cell growth and metastatic behavior (36). Moreover, MDSCs enhance PD-L1 expression, resulting in diminished natural killer cell and T lymphocyte function while reinforcing the immunosuppressive characteristics of the tumor niche (37). As the most efficient professional antigen-presenting cells, DCs play a pivotal role in priming naïve T cells and orchestrating adaptive immunity (38). Within GBM, overexpression of macrophage migration inhibitory factor (MIF) facilitates disease progression by triggering autophagic processes and impairing DC-mediated immune recognition (39). Furthermore, the oncogenic protein annexin A1, which is highly expressed in glioblastoma, disrupts DC differentiation via NF-κB-dependent mechanisms. This leads to elevated IL-8 secretion and p65 phosphorylation, collectively promoting tumor immune evasion (40). Additionally, tumor-derived PGE2 contributes to immune suppression by inhibiting dendritic cell maturation through EP2 and EP4 receptor signaling. Activation of these G protein–coupled receptors elevates intracellular cAMP levels and downstream PKA activity, thereby reducing MHC class II expression and costimulatory molecules such as CD80 and CD86 (41). This impaired maturation limits the antigen-presenting capacity of DCs and blunts the priming of cytotoxic T lymphocytes, further facilitating immune evasion in the GBM microenvironment.
2.3 Microglia
Microglia serve as the principal immunocompetent cells within the central nervous system, maintaining a quiescent surveillance state characterized by branched processes during homeostasis, yet rapidly transitioning to an activated phenotype upon encountering pathological triggers to execute immunoprotective functions (42). Within glioma ecosystems, these neural immune effectors migrate toward tumor masses guided by chemoattractant molecules including CCL2, subsequently secreting an array of soluble mediators such as interleukin 6, transforming growth factor beta, and vascular endothelial growth factor that collectively promote neoplastic invasion and expansion (43). The immune architecture of gliomas emerges from dynamic intercellular communication among diverse leukocyte populations. Of particular significance, tumor infiltrating macrophages generate immunosuppressive factors including transforming growth factor beta and interleukin 10 that simultaneously dampen CD8+ T cell effector mechanisms while stimulating regulatory T cell clonal expansion, thereby cultivating an immunotolerant microenvironment (12). Additionally, these macrophage populations overexpress coinhibitory molecules including programmed death ligand 1 that directly compromise T cell antitumor capacity. The synergistic action of immunosuppressive mediators originating from both neoplastic cells and tumor associated macrophages promotes T cell hyporesponsiveness, ultimately subverting antitumor immunity during glioma pathogenesis (44). The programmed death 1 programmed death ligand 1 axis constitutes a fundamental pathway facilitating glioma immune evasion. Neoplastic cells increase programmed death ligand 1 surface expression to engage programmed death 1 receptors on T lymphocytes, resulting in functional inhibition and progressive exhaustion that enables tumor immune escape. This immunosuppressive cascade becomes further intensified through the combined action of inhibitory cytokines including transforming growth factor beta and interleukin 10 together with regulatory immune cell populations such as tumor associated macrophages and regulatory T cells, ultimately generating a sophisticated and multilayered immune suppression network (45).
2.4 Adaptive immunity
Within the tumor microenvironment of GBM, CD4+ and CD8+ T lymphocytes constitute the dominant adaptive immune cell subsets, comprising roughly 5% of all CD45+ leukocytic infiltrates (46). Notably, GBM with wild-type isocitrate dehydrogenase (IDH) status demonstrate greater T cell infiltration compared to their IDH-mutant counterparts (47). Despite their presence, these T cells often undergo functional exhaustion, primarily driven by chronic exposure to tumor-associated antigens. This exhausted phenotype is characterized by reduced proliferation, compromised cytotoxic activity, and elevated expression of immune-inhibitory molecules, including PD-1 and CTLA-4 (48). Furthermore, immunosuppression within GBM is amplified by CD4+CD25+FoxP3+ regulatory T cells (Tregs), which actively suppress antitumor immunity (49). While cytotoxic T cells retain intrinsic tumoricidal capacity, their efficacy is substantially hindered in the GBM TME due to heightened immune checkpoint signaling and the prevalence of immunosuppressive cellular populations (50). Despite constituting a relatively small fraction of immune infiltrates in GBM, B lymphocytes play a pivotal role in modulating tumor biology and treatment outcomes (51). The B cell compartment within GBM includes both immunoregulatory B cells (Bregs) that suppress immune responses and conventional B lymphocytes capable of antigen presentation, which can amplify T cell activation (52, 53). These cells mediate immunosuppressive effects primarily through the production of IL-10 and TGF-β, cytokines that impair the cytotoxic function of T cells and NK cells while simultaneously promoting processes associated with neural development and tumor infiltration (54). Additionally, B cells contribute to tumor vascularization by secreting angiogenic factors, including VEGF, CXCL12, and CXCL13. These molecules foster the formation of new blood vessels, thereby enhancing oxygen and nutrient delivery to support tumor growth (Table 1) (54).
Cell typeMarkersKey functionImmunosuppressive mechanismsTreatmentTAMsMicroglia (TMEM119+, P2RY12+); BMDMs (CCR2+, CD45+high)ECM remodeling, angiogenesis, PD-L1 induction, immunosuppressionIL-10, TGF-β, PD-L1, M2 polarization, STAT3 activationCSF1R inhibition, PD-L1 blockade, TAM reprogrammingT Lymphocytes (CD4+, CD8+, Tregs)CD3+, CD4+/CD8+; Tregs: CD25+FoxP3+Cytotoxicity, immunoregulation, tolerance inductionPD-1, CTLA-4 upregulation, Treg suppressionCheckpoint inhibitors, Treg depletionNeutrophilsCD11b+Ly6G+Early anti-tumor effects; later promote angiogenesis, invasionReprogramming by TME signals; cytokine releaseCXCR2 blockade, inhibit late-phase polarizationB CellsCD19+, CD20+; BregsIL-10 and VEGF secretion, antigen presentationTGF-β and IL-10 suppress effector immunity; promote angiogenesisAnti-VEGF/IL-10 therapy, B cell modulationNK CellsCD56+CD3-; NKp46+, NKG2D+, CD16+Kill tumor & GSCs, mediate ADCCDownregulation of activating ligands; suppression by TGF-β, IL-10TGF-β blockade, ligand expression restorationMDSCsCD11b+, Gr1+, HLA-DR-Suppress T/NK cells, secrete IL-6, promote angiogenesisSTAT3 activation, PD-L1/IL-6 productionSTAT3/IL-6 inhibition, anti-MDSC therapiesDendritic CellsCD11c+MHC-II+, CD80+, CD86+Antigen presentation, T cell primingMIF, Annexin A1 impair maturation, induce IL-8 and p65 activationDC vaccines, maturation stimulantsMicrogliaTMEM119+, Iba1+, CX3CR1+Secrete IL-6, VEGF, TGF-β; support invasion, regulate local immunityPromote Treg expansion, PD-L1 expression, release immunosuppressive cytokinesReprogramming, M1 activation, checkpoint inhibition
Immune cells in the GBM microenvironment.
3 Biological mechanisms underlying the GBM microenvironment
Glioblastoma represents a complex biological system where diverse cell types and molecular mediators interact through intricate signaling networks, driving malignancy and therapeutic resistance. A hallmark of GBM is aberrant angiogenesis, primarily driven by elevated expression of angiogenic mediators such as VEGF, bFGF, HGF, PDGF, TGF-β, MMPs, and angiopoietins. These factors are upregulated through oncogene activation, tumor suppressor loss, and hypoxia-induced stress responses (55, 56). FGFRs promote neovascularization via activation of PI3K/AKT/mTOR and c-JUN/p38-MAPK/STAT3/NF-κB pathways, facilitating tumor growth and vascular development (57, 58). The extracellular matrix (ECM) supports vascular expansion, tumor infiltration, and resistance to therapy (59). Key ECM components include fibronectin-C, which enhances cellular invasiveness (60), fibronectin, which contributes to chemoresistance (61), Fibulin-3, which activates Notch/NF-κB and induces IL-6 secretion from TAMs through integrin αvβ3/FAK signaling (62), and hyaluronic acid, which accelerates cell migration (61). Metabolic reprogramming and immune regulation are tightly linked in GBM. Lactate accumulation acidifies the microenvironment, impairing cytotoxic T and NK cell function while expanding Tregs and MDSCs (63). Beyond acidification, lactate activates GPR81 on TAMs, suppressing NF-κB signaling and promoting an immunosuppressive TAM phenotype. TAM-derived IL-10 and TGF-β drive metabolic shifts toward glycolysis and lipid synthesis, enhancing tumor cell survival (64). Targeting lactate reduces immune evasion and genomic repair via XRCC1 lactylation blockade, improving treatment efficacy (65). Moreover, enzymes such as IDO co-express with immune checkpoints like PD-L1, reinforcing immune tolerance (66). Cytokines and chemokines mediate bidirectional communication between GBM cells and immune elements. IL-6 promotes tumor proliferation, while TGF-β and IL-10 enhance Treg-mediated immunosuppression (67). Tumor-derived exosomes carrying PD-L1 and proteases reshape the microenvironment and facilitate metastasis (68). Hypoxia activates HIFs, promoting cell proliferation, angiogenesis, and metabolic adaptation (69), while also impairing NK and effector T cell function and enriching immunosuppressive cells such as Tregs and MDSCs (70, 71). The convergence of vascular remodeling, ECM dynamics, metabolic adaptation, and immune evasion underscores the complexity of the GBM niche and its role in therapy resistance.
4 Tumor microenvironment-targeted therapies in GBM4.1 Immune checkpoint inhibitors
Immunological checkpoint molecules, primarily expressed on immune effector cells—especially T lymphocytes—are essential for maintaining self-tolerance and preventing autoimmune responses. However, malignant cells frequently hijack these regulatory pathways to evade immune-mediated elimination (72). Checkpoint blockade therapeutics reinvigorate the cytotoxic potential of T cells. Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), the initial clinically validated immune checkpoint, attenuates T cell stimulation by outcompeting CD28 for binding to CD80/CD86 ligands on antigen-presenting cells. Clinical evaluation of ipilimumab, an antagonist of CTLA-4, in patients with glioblastoma revealed no improvement in outcomes compared to temozolomide in Phase II clinical trials (73). The PD-1/PD-L1 signaling axis mediates T cell suppression, with elevated PD-L1 expression serving as a negative prognostic indicator (74–76). CD47 interaction with SIRPα on phagocytic cells prevents tumor cell engulfment, thereby driving disease recurrence; disruption of this molecular interaction could potentiate checkpoint immunotherapy (77, 78). Elevated TIM-3 levels in GBM correlate with enhanced tumor aggressiveness (79, 80), whereas indoleamine 2,3-dioxygenase (IDO) suppresses cytotoxic lymphocyte function and shows increased activity in GBM specimens (81). Pharmacological inhibition of IDO, such as through the use of epacadostat, has shown promising results in preclinical studies, particularly when combined with radiotherapy or anti-PD-1 immunotherapy (82). The limited clinical efficacy of immune checkpoint inhibitors in glioblastoma is multifactorial. One contributing mechanism is the immunosuppressive tumor microenvironment, characterized by the secretion of interleukin-10 and transforming growth factor-beta by M2-polarized tumor-associated macrophages and myeloid-derived suppressor cells, which collectively suppress cytotoxic immune responses (83). Additionally, the inherently low tumor mutational burden in glioblastoma results in a scarcity of immunogenic neoantigens, thereby reducing the likelihood of effective immune recognition (84). Furthermore, the structural and functional integrity of the blood-brain barrier restricts the intratumoral delivery of immunotherapeutic agents, further limiting clinical benefit. Recent studies have demonstrated that galectin-9, secreted by glioblastoma stem-like cells, engages the TIM-3 receptor on Th1 lymphocytes, contributing to their functional exhaustion and promoting immune evasion (85). This interaction activates intracellular apoptotic signaling cascades, leading to Th1 cell apoptosis, while concurrently suppressing the production of key cytokines such as interleukin-2 and interferon-gamma. The resulting impairment of Th1-mediated immune responses facilitates the establishment of an immunosuppressive tumor microenvironment conducive to glioblastoma progression (Figure 1) (86).
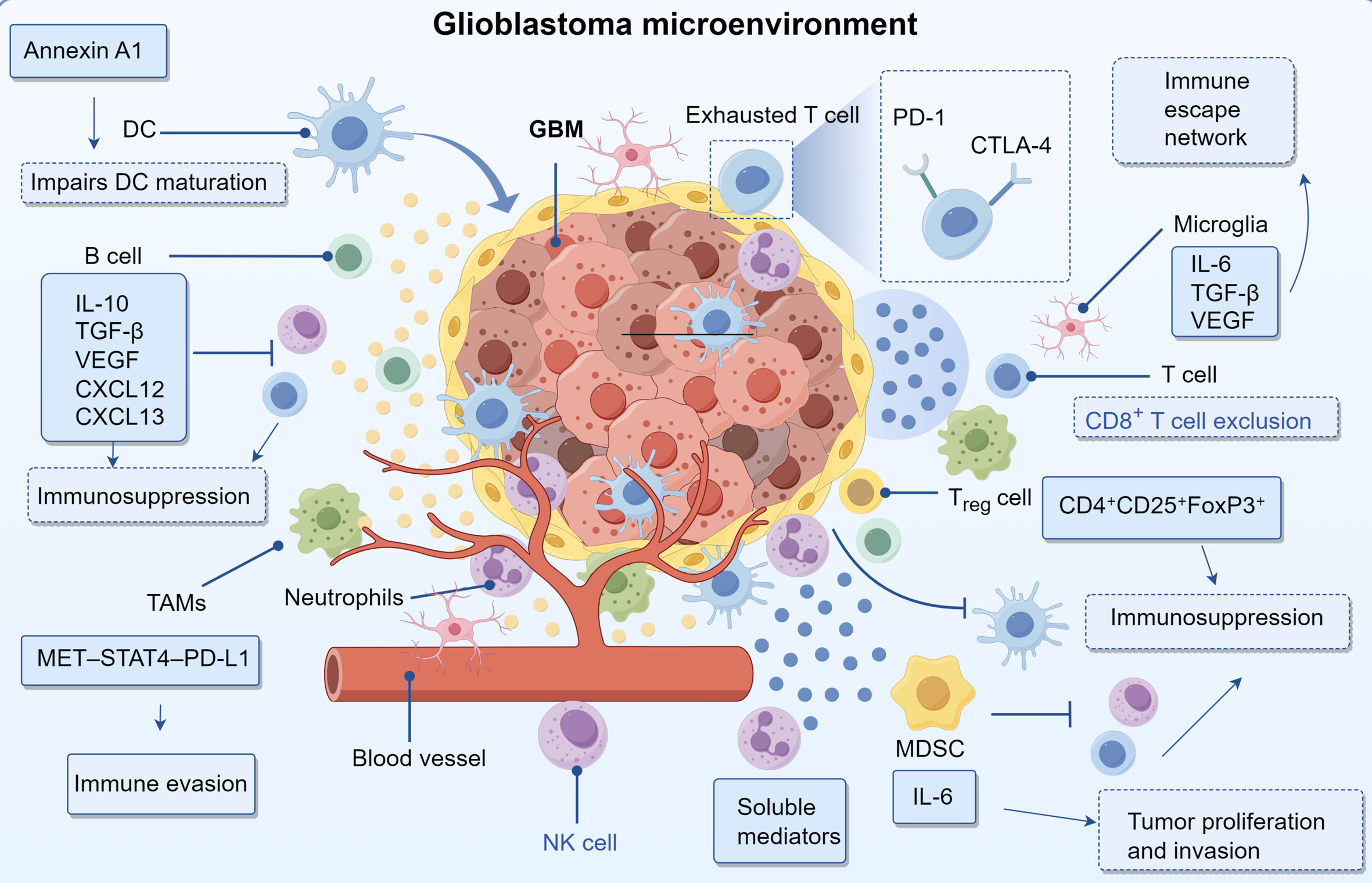
Mechanism of immune cells in GBM progression.
4.2 Chimeric antigen receptor T cell therapy
As a groundbreaking form of immunotherapy, CAR-T cell therapy involves genetically engineering autologous T lymphocytes to express synthetic receptors that recognize tumor-associated antigens independently of MHC restriction. Despite this innovation, its efficacy against GBM remains limited, with median overall survival (mOS) reaching only 8 months for EGFRvIII-targeted therapy and 11.1 months for HER2-directed approaches, underscoring challenges posed by tumor invasiveness and immune evasion (87). Key challenges include: (1) antigenic heterogeneity enabling immune escape; (2) an immunosuppressive microenvironment enriched with MDSCs and Tregs; (3) poor CAR-T cell trafficking across the BBB; and (4) antigen loss through clonal selection. Recent studies demonstrate that trogocytosis mediates CAR molecule transfer to tumor cells, a resistance mechanism modulated by antigen density, receptor affinity, and lipid metabolism (87). Current research focuses on optimizing CAR activation thresholds to adapt to heterogeneous antigen expression.
4.3 Oncolytic virotherapy
A novel immunotherapeutic approach for GBM involves the use of oncolytic viruses, which are genetically modified to preferentially infect and propagate within malignant cells, leading to their destruction (88). This process triggers the release of danger-associated molecular patterns (DAMPs) along with proinflammatory cytokines, transforming immunologically inert (“cold”) tumors into immunologically active (“hot”) lesions (89). Among the earliest viruses adapted for GBM therapy is herpes simplex virus (HSV), with modified variants including HSV-1716, G207, and G47Δ demonstrating favorable safety profiles and selective tumor tropism in preclinical and early clinical studies (90, 91). Additionally, PVSRIPO—a recombinant poliovirus-rhinovirus chimera—has been developed to target CD155, a receptor frequently upregulated in GBM (92). In a Phase I clinical trial (NCT01491893), intratumoral administration of PVSRIPO in recurrent GBM patients was well tolerated, yielding a median overall survival (mOS) of 12.5 months—exceeding the historical benchmark of 11.3 months. Furthermore, survival rates at 24 and 36 months reached 21%, significantly higher than those observed in control groups (14% and 4%, respectively) (93). Ongoing investigations include a Phase II monotherapy trial (NCT02986178) as well as combination studies incorporating immune checkpoint inhibitors such as atezolizumab (anti–PD-L1) or pembrolizumab (anti–PD-1) (NCT03973879, NCT04479241), aiming to evaluate potential synergistic effects.
4.4 Cancer vaccines
Designed to elicit adaptive immunity targeting tumor-associated or tumor-specific antigens, cancer vaccines represent a promising strategy for GBM treatment (94). These immunogens can originate from endogenous tumor-derived proteins or exogenous pathogens such as cytomegalovirus (CMV) (95, 96). Some formulations incorporate predefined antigens, whereas others rely on antigen-presenting cell (APC) activation to process unidentified tumor epitopes (97). Upon delivery, APCs prime T cells, which subsequently infiltrate malignant tissue, triggering cytotoxic activity and potentially generating durable immunological memory. Current vaccine modalities explored for GBM comprise peptide-based formulations, dendritic cell (DC)-based vaccines, nucleic acid (DNA/RNA) vaccines, and viral vector-delivered immunogens. As specialized APCs, DCs internalize tumor antigens and traffic to lymphoid organs to stimulate T cell activation (98, 99). DC vaccination entails isolating patient-derived DCs, pulsing them with tumor antigens, and reinfusing them to induce tumor-specific T cell responses (100). Preclinical studies demonstrated that DC vaccines incorporating EGFRvIII-transfected glioma cells elicited potent antitumor immunity and increased survival. A Phase II clinical trial assessing DC vaccination in newly diagnosed GBM patients reported enhanced modest overall survival benefits (101). Pooled analyses indicate that DC-based immunization substantially improves 1- and 2-year OS rates in treatment-naïve GBM cases (102). Furthermore, autologous tumor lysate-loaded DC vaccines conferred survival advantages in both newly diagnosed and recurrent GBM relative to conventional therapy, particularly benefiting MGMT-methylated subgroups (103).
5 Conclusion
Glioblastoma (GBM) presents a formidable therapeutic challenge due to its profoundly immunosuppressive TME, which is predominantly shaped by innate immune mechanisms. TAMs, MDSCs, and microglia establish an immunosuppressive niche through multiple mechanisms, including cytokine secretion (IL-10, TGF-β), metabolic reprogramming, and immune checkpoint upregulation. These cells not only facilitate tumor progression but also actively suppress adaptive immune responses, rendering conventional immunotherapies largely ineffective. The complexity of these interactions underscores the need for novel strategies that specifically target innate immune pathways to disrupt the immunosuppressive network and restore antitumor immunity.
Emerging therapeutic approaches, such as TAM repolarization, MDSC depletion, and NK cell activation, show promise in reshaping the GBM microenvironment. However, their full potential will likely be realized only when combined with other modalities, including immune checkpoint blockade, metabolic interventions, and precision targeting. Future research should focus on elucidating the intricate crosstalk between innate and adaptive immunity while developing integrated treatment strategies that simultaneously overcome immunosuppression and enhance tumor-specific immune responses. A paradigm shift toward innate immune-focused therapies, within a comprehensive multimodal framework, may finally break the therapeutic impasse in this.
StatementsAuthor contributions
WeZ: Writing – original draft. WaZ: Writing – original draft. HW: Writing – review & editing. XH: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research and/or publication of this article.
Conflict of interest
The authors declare that no competing financial interests or commercial relationships have influenced the research presented herein.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
YuKHuYWuFGuoQQianZHuWet al. Surveying brain tumor heterogeneity by single-cell RNA-sequencing of multi-sector biopsies. Natl Sci Rev. (2020) 7:1306–18. doi: 10.1093/nsr/nwaa099
SahaSBhatAKukalSPhalakMKumarS. Spatial heterogeneity in glioblastoma: Decoding the role of perfusion. Biochim Biophys Acta Rev Cancer. (2025) 1880:189383. doi: 10.1016/j.bbcan.2025.189383
WenNXiaoXLuHChenQHeGQianZet al. Immuno-oncological interactions between meningeal lymphatics and glioblastoma: from mechanisms to therapies. Theranostics. (2025) 15:6983–7000. doi: 10.7150/thno.111972
JiangTNamDHRamZPoonWSWangJBoldbaatarDet al. Clinical practice guidelines for the management of adult diffuse gliomas. Cancer Lett. (2021) 499:60–72. doi: 10.1016/j.canlet.2020.10.050
KennedyLBSalamaAKS. A review of cancer immunotherapy toxicity. CA Cancer J Clin. (2020) 70:86–104. doi: 10.3322/caac.21596
ReardonDABrandesAAOmuroAMulhollandPLimMWickAet al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: the checkMate 143 phase 3 randomized clinical trial. JAMA Oncol. (2020) 6:1003–10. doi: 10.1001/jamaoncol.2020.1024
OmuroABrandesAACarpentierAFIdbahAReardonDACloughesyTet al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: an international randomized phase III trial. Neuro Oncol. (2023) 25:123–34. doi: 10.1093/neuonc/noac099
LimMWellerMIdbahASteinbachJFinocchiaroGRavalRRet al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. (2022) 24:1935–49. doi: 10.1093/neuonc/noac116
MedikondaRAbikenariMSchonfeldELimM. The metabolic orchestration of immune evasion in glioblastoma: from molecular perspectives to therapeutic vulnerabilities. Cancers. (2025) 17:1881. doi: 10.3390/cancers17111881
KarbhariNFrechetteKMBurnsTCParneyIFCampianJLBreenWGet al. Immunotherapy for high-grade gliomas. Cancers. (2025) 17:1849. doi: 10.3390/cancers17111849
AlemánORQuinteroJCCamacho-ArroyoI. The language of glioblastoma: A tale of cytokines and sex hormones communication. Neuro Oncol Adv. (2025) 7:vdaf017. doi: 10.1093/noajnl/vdaf017
LinHLiuCHuAZhangDYangHMaoY. Understanding the immunosuppressive microenvironment of glioma: mechanistic insights and clinical perspectives. J Hematol Oncol. (2024) 17:31. doi: 10.1186/s13045-024-01544-7
ZhaoWZhangZXieMDingFZhengXSunSet al. Exploring tumor-associated macrophages in glioblastoma: from diversity to therapy. NPJ Precis Oncol. (2025) 9:126. doi: 10.1038/s41698-025-00920-x
MatsuzakiHPanCKomoharaYYamadaRYanoHFujiwaraYet al. The roles of glioma-associated macrophages/microglia and potential targets for anti-glioma therapy. Immunol Med. (2025) 48:24–32. doi: 10.1080/25785826.2024.2411035
XuCXiaoMLiXXinLSongJZhanQet al. Origin, activation, and targeted therapy of glioma-associated macrophages. Front Immunol. (2022) 13:974996. doi: 10.3389/fimmu.2022.974996
PomboAAScheyltjensILodiFMessiaenJAntoranzADuerinckJet al. Single-cell profiling of myeloid cells in glioblastoma across species and disease stage. Nat Neurosci. (2021) 24:595–610. doi: 10.1038/s41593-020-00789-y
KhanFPangLDuntermanMLesniakMSHeimbergerABChenP. Macrophages and microglia in glioblastoma: heterogeneity, plasticity, and therapy. J Clin Invest. (2023) 133:e163446. doi: 10.1172/JCI163446
ChenZRossJLHambardzumyanD. Intravital 2-photon imaging reveals distinct morphology and infiltrative properties of glioblastoma-associated macrophages. Proc Natl Acad Sci U.S.A. (2019) 116:14254–9. doi: 10.1073/pnas.1902366116
ZhouWKeSQHuangZFlavahanWFangXPaulJet al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes Malignant growth. Nat Cell Biol. (2015) 17:170–82. doi: 10.1038/ncb3090
HuX. Microglia/macrophage polarization: fantasy or evidence of functional diversity? J Cereb Blood Flow Metab. (2020) 40:S134–6. doi: 10.1177/0271678X20963405
ChengNBaiXShuYAhmadOShenP. Targeting tumor-associated macrophages as an antitumor strategy. J Biochem Pharmacol. (2021) 183:114354. doi: 10.1016/j.bcp.2020.114354
Shapouri-MoghaddamAMohammadianSVaziniHTaghadosiMEsmaeiliSAMardaniFet al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
LinCWangXXuC. Glioma-associated microglia/macrophages (GAMs) in glioblastoma: immune function in the tumor microenvironment and implications for immunotherapy. Front Immunol. (2023) 14:1123853. doi: 10.3389/fimmu.2023.1123853
WangQWSunLHZhangYWangZZhaoZWangZLet al. MET overexpression contributes to STAT4-PD-L1 signaling activation associated with tumor-associated, macrophages-mediated immunosuppression in primary glioblastomas. J Immunother Cancer. (2021) 9:e002451. doi: 10.1136/jitc-2021-002451
ZhouJPeiXYangYWangZGaoWYeRet al. Orphan nuclear receptor TLX promotes immunosuppression via its transcriptional activation of PD-L1 in glioma. J Immunother Cancer. (2021) 9:e001937. doi: 10.1136/jitc-2020-001937
ZhanYQiaoWYiBYangXLiMSunLet al. Dual role of pseudogene TMEM198B in promoting lipid metabolism and immune escape of glioma cells. Oncogene. (2022) 41:4512–23. doi: 10.1038/s41388-022-02445-0
SunCWangSMaZZhouJDingZYuanGPanY. Neutrophils in glioma microenvironment: from immune function to immunotherapy. Front Immunol. (2024) 15:1393173. doi: 10.3389/fimmu.2024.1393173
MassaraMPersicoPBonavitaOMollicaPoetaLocatiMSimonelliMet al. Neutrophils in gliomas. Front Immunol. (2017) 8:1349. doi: 10.3389/fimmu.2017.01349
WangGWangJNiuCZhaoYWuPNiuCet al. Neutrophils: New critical regulators of glioma. Front Immunol. (2022) 13:927233. doi: 10.3389/fimmu.2022.927233
VivierETomaselloEBaratinMWalzerTUgoliniS. Functions of natural killer cells. Nat Immunol. (2008) 9:503–10. doi: 10.1038/ni1582
FujitaMKohanbashGFellows-MayleWHamiltonRLKomoharaYDeckerSAet al. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. J Cancer Res. (2011) 71:2664–74. doi: 10.1158/0008-5472.CAN-10-3055
BreznikBKoMWTseCChenPCSenjorEMajcBHabičAet al. Infiltrating natural killer cells bind, lyse and increase chemotherapy efficacy in glioblastoma stem-like tumorospheres. Commun Biol. (2022) 5:436. doi: 10.1038/s42003-022-03402-z
ShanleyMDaherMDouJLiSBasarRRafeiHet al. Interleukin-21 engineering enhances NK cell activity against glioblastoma via CEBPD. J Cancer Cell. (2024) 42:1450–1466.e11. doi: 10.1016/j.ccell.2024.07.007
FaresJDavisZBRechbergerJSTollSASchwartzJDDanielsDJet al. Advances in NK cell therapy for brain tumors. NPJ Precis Oncol. (2023) 7:17. doi: 10.1038/s41698-023-00356-1
BayikDZhouYParkCHongCVailDSilverDJet al. Myeloid-derived suppressor cell subsets drive glioblastoma growth in a sex-specific manner. Cancer Discov. (2020) 10:1210–25. doi: 10.1158/2159-8290.CD-19-1355
GeYChengDJiaQXiongHZhangJ. Mechanisms underlying the role of myeloid-derived suppressor cells in clinical diseases: good or bad. Immune Netw. (2021) 21:e21. doi: 10.4110/in.2021.21.e21
EichlerAFChungEKodackDPLoefflerJSFukumuraDJainRK. The biology of brain metastases-translation to new therapies. Nat Rev Clin Oncol. (2011) 8:344–56. doi: 10.1038/nrclinonc.2011.58
SunYChenYTaoXZhangWWangXWangXet al. INPP4B inhibits glioma cell proliferation and immune escape via inhibition of the PI3K/AKT signaling pathway. Front Oncol. (2022) 12:985337. doi: 10.3389/fonc.2022.985337
PanDSFengSZCaoPLiJJ. Endothelin B receptor promotes the proliferation and immune escape of Malignant gliomas. Antif Cells Nanomed Biotechnol. (2018) 46:1230–5. doi: 10.1080/21691401.2017.1366336
MurphyTLMurphyKM. Dendritic cells in cancer immunology. J Cell Mol Immunol. (2022) 19:3–13. doi: 10.1038/s41422-021-00471-5
Cuenca-EscalonaJFlórez-GrauGvan den DriesKCambiAde VriesIJM. PGE2-EP4 signaling steers cDC2 maturation toward the induction of suppressive T-cell responses. Eur J Immunol. (2024) 54:e2350770. doi: 10.1002/eji.202350770
HanischUKKettenmannH. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. (2007) 10:1387–94. doi: 10.1038/nn1997
KioiMVogelHSchultzGHoffmanRMHarshGRBrownJM. Inhibition of vasculogenesis, but not angiogenesis, prevents the recurrence of glioblastoma after irradiation in mice. J Clin Invest. (2010) 120:694–705. doi: 10.1172/JCI40283
ZhangHLiuLLiuJDangPHuSYuanWet al. Roles of tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for solid cancers. Mol Cancer. (2023) 22:58. doi: 10.1186/s12943-023-01725-x
WangHZhouHXuJLuYJiXYaoYet al. Different T-cell subsets in glioblastoma multiforme and targeted immunotherapy. Cancer Lett. (2021) 496:134–43. doi: 10.1016/j.canlet.2020.09.028
MaasSLNStichelDHielscherTSieversPBerghoffASSchrimpfDet al. Integrated molecular-morphologic meningioma classification: a multicenter retrospective analysis, retrospectively and prospectively validated. J Clin Oncol. (2021) 39:3839–52. doi: 10.1200/JCO.21.00784
WischnewskiVMaasRRAruffoPGSoukupKGallettiGKorneteMet al. Phenotypic diversity of T cells in human primary and metastatic brain tumors revealed by multiomic interrogation. Nat Cancer. (2023) 4:908–24. doi: 10.1038/s43018-023-00566-3
WherryEJKurachiM. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. (2015) 15:486–99. doi: 10.1038/nri3862
El AndaloussiALesniakMS. An increase in CD4+ CD25+ FOXP3+ regulatory T cells in tumor-infiltrating lymphocytes of human glioblastoma multiforme. Neuro Oncol. (2006) 8:234–43. doi: 10.1215/15228517-2006-006
QinZHuangYLiZPanGZhengLXiaoXet al. Glioblastoma vascular plasticity limits effector T-cell infiltration and is blocked by cAMP activation. Cancer Immunol Res. (2023) 11:1351–66. doi: 10.1158/2326-6066.CIR-22-0872
JainRWYongVW. B cells in central nervous system disease: diversity, locations and pathophysiology. Nat Rev Immunol. (2022) 22:513–24. doi: 10.1038/s41577-021-00652-6
HouDWanHKatzJLWangSCastroBAVazquez-CervantesGIet al. Antigen-presenting B cells promote TCF-1+ PD1– stem-like CD8+ T-cell proliferation in glioblastoma. Front Immunol. (2024) 14:1295218. doi: 10.3389/fimmu.2023.1295218
Lee-ChangCRashidiAMiskaJZhangPPituchKCHouDet al. Myeloid-derived suppressive cells promote B cell-mediated immunosuppression via transfer of PD-L1 in glioblastoma. Cancer Immunol Res. (2019) 7:1928–43. doi: 10.1158/2326-6066.CIR-19-0240
ShonkaNPiaoYGilbertMYungAChangSDeAngelisLMet al. Cytokines associated with toxicity in the treatment of recurrent glioblastoma with aflibercept. J Target Oncol. (2013) 8:117–25. doi: 10.1007/s11523-013-0254-0
SiemannDWChaplinDJHorsmanMR. Realizing the potential of vascular targeted therapy: the rationale for combining vascular disrupting agents and anti-angiogenic agents to treat cancer. Cancer Invest. (2017) 35:519–34. doi: 10.1080/07357907.2017.1364745
BatchelorTTReardonDAde GrootJFWickWWellerM. Antiangiogenic therapy for glioblastoma: current status and future prospects. Clin Cancer Res. (2014) 20:5612–9. doi: 10.1158/1078-0432.CCR-14-0834
OnishiMKurozumiKIchikawaTDateI. Mechanisms of tumor development and anti-angiogenic therapy in glioblastoma multiforme. Neurol Med Chir (Tokyo). (2013) 53:755–65. doi: 10.2176/nmc.ra.2013-0200
SoomanLFreyhultEJaiswalANavaniSEdqvistPHPonténFet al. FGF2 as a potential prognostic biomarker for proneural glioma patients. Acta Oncol. (2015) 54:385–94. doi: 10.3109/0284186X.2014.951492
MohiuddinEWakimotoH. Extracellular matrix in glioblastoma: opportunities for emerging therapeutic approaches. Am J Cancer Res. (2021) 11:3742–54.
BrösickeNFaissnerA. Role of tenascins in the ECM of gliomas. Cell Adh Migr. (2015) 9:131–40. doi: 10.1080/19336918.2014.1000071
YuQXueYLiuJXiZLiZLiuY. Fibronectin promotes the Malignancy of glioma stem-like cells via modulation of cell adhesion, differentiation, proliferation and chemoresistance. Front Mol Neurosci. (2018) 11:130. doi: 10.3389/fnmol.2018.00130
NandhuMSBeheraPBhaskaranVLongoSLBarrera-ArenasLMSenguptaSet al. Development of a function-blocking antibody against fibulin-3 as a targeted reagent for glioblastoma. Clin Cancer Res. (2018) 24:821–33. doi: 10.1158/1078-0432.CCR-17-1628
BrandASingerKKoehlGEKolitzusMSchoenhammerGThielAet al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. (2016) 24:657–71. doi: 10.1016/j.cmet.2016.08.011
QuailDFJoyceJA. The Microenvironmental landscape of brain tumors. J Cancer Cell. (2017) 31:326–41. doi: 10.1016/j.ccell.2017.02.009
LiGWangDZhaiYPanCZhangJWangCet al. Glycometabolic reprogramming-induced XRCC1 lactylation confers therapeutic resistance in ALDH1A3-overexpressing glioblastoma. Cell Metab. (2024) 36:1696–1710.e10. doi: 10.1016/j.cmet.2024.07.011
PlattenMWickWVan den EyndeBJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res. (2012) 72:5435–40. doi: 10.1158/0008-5472.CAN-12-0569
WangQHuBHuXKimHSquatritoMScarpaceLet al. Tumor evolution of glioma-intrinsic gene expression subtypes associates with immunological changes in the microenvironment. J Cancer Cell. (2017) 32:42–56.e6. doi: 10.1016/j.ccell.2017.06.003
OstiDDel BeneMRappaGSantosMMataforaVRichichiCet al. Clinical significance of extracellular vesicles in plasma from glioblastoma patients. Clin Cancer Res. (2019) 25:266–76. doi: 10.1158/1078-0432.CCR-18-1941
KaurBKhwajaFWSeversonEAMathenySLBratDJVan MeirEG. Hypoxia and the hypoxia-inducible-factor pathway in glioma growth and angiogenesis. Neuro Oncol. (2005) 7:134–53. doi: 10.1215/15228517-2005-112
PalazónAAragonésJMorales-KastresanaAde LandázuriMOMeleroI. Molecular pathways: hypoxia response in immune cells fighting or promoting cancer. Clin Cancer Res. (2012) 18:1207–13. doi: 10.1158/1078-0432.CCR-11-1591
NomanMZHasmimMMessaiYTerrySKiedaCJanjiBet al. Hypoxia: a key player in antitumor immune response. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol Cell Physiol. (2015) 309:C569–79. doi: 10.1152/ajpcell.00207.2015
GaikwadSAgrawalMYKaushikIRamachandranSSrivastavaSK. Immune checkpoint proteins: signaling mechanisms and molecular interactions in cancer immunotherapy. Semin Cancer Biol. (2022) 86:137–50. doi: 10.1016/j.semcancer.2022.03.014
BrownNFNgSMBrooksCCouttsTHolmesJRobertsCet al. A phase II open label, randomised study of ipilimumab with temozolomide versus temozolomide alone after surgery and chemoradiotherapy in patients with recently diagnosed glioblastoma: the Ipi-Glio trial protocol. BMC Cancer. (2020) 20:198. doi: 10.1186/s12885-020-6624-y
FranciscoLMSalinasVHBrownKEVanguriVKFreemanGJKuchrooVKet al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. (2009) 206:3015–29. doi: 10.1084/jem.20090847
NoonanKEWeiJYaghiNKHuangNKongLYGabrusiewiczKet al. PD-L1 expression in glioblastoma is associated with the presence of CD8+ tumor-infiltrating lymphocytes and prognosis. J Neuro Oncol. (2016) 18:195–205. doi: 10.1002/ponc.4106
BerghoffASKieselBWidhalmGRajkyORickenGWöhrerAet al. Programmed death ligand 1 expression and tumor-infiltrating lymphocytes in glioblastoma. Neuro Oncol. (2015) 17:1064–75. doi: 10.1093/neuonc/nov172
GholaminSMitraSSFerozeAHLiuJKahnSAZhangMet al. Disrupting the CD47–SIRPα anti-phagocytic axis by a humanized anti-CD47 antibody is an efficacious treatment for Malignant pediatric brain tumors. Sci Transl Med. (2017) 9:eaaf2968. doi: 10.1126/scitranslmed.aaf2968
ZhangMHutterGKahnSAAzadTDGholaminSXuCYet al. Anti-CD47 treatment stimulates phagocytosis of glioblastoma by M1 and M2 polarized macrophages and promotes M1 polarized macrophages in vivo. J Natl Cancer Inst. (2016) 114:e0153550. doi: 10.1371/journal.pone.0153550
DasMZhuCKuchrooVK. Tim-3 and its role in regulating anti-tumor immunity. J Immunol Rev. (2017) 276:97–111. doi: 10.1111/imr.12520
HanSFengSXuLShiWWangXWangHet al. Tim-3 on peripheral CD4+ and CD8+ T cells is involved in the development of glioma. DNA Cell Biol. (2014) 33:245–50. doi: 10.1089/dna.2013.2306
CheongJEEkkatiASunL. A patent review of IDO1 inhibitors for cancer. Expert Opin Ther Pat. (2018) 28:317–30. doi: 10.1080/13543776.2018.1441249
HaniharaMKawatakiTOh-OkaKMitsukaKNakaoAKinouchiH. Synergistic antitumor effect with indoleamine 2,3-dioxygenase inhibition and temozolomide in a murine glioma model. J Neurosurg. (2016) 124:1594–601. doi: 10.3171/2015.JNS149101
De LeoAUgoliniAYuXScirocchiFScocozzaDPeixotoBet al. Glucose-driven histone lactylation promotes the immunosuppressive activity of monocyte-derived macrophages in glioblastoma. Immunity. (2024) 57:1105–1123.e8. doi: 10.1016/j.immuni.2024.04.006
RodriguezSMBTataranuLGKamelATurliucSRizeaREDricuA. Glioblastoma and immune checkpoint inhibitors: A glance at available treatment options and future directions. Int J Mol Sci. (2024) 25:10765. doi: 10.3390/ijms251910765
KandelSAdhikaryPLiGChengK. The TIM3/Gal9 signaling pathway: An emerging target for cancer immunotherapy. Cancer Lett. (2021) 510:67–78. doi: 10.1016/j.canlet.2021.04.011
FengXLSuGWuQHJiaQZhangZC. Research progress of galectins in glioma. Discover Oncol. (2025) 16:1003. doi: 10.1007/s12672-025-02318-4
ZhaiYDuYLiGYuMHuHPanCet al. Trogoctyosis of CAR molecule regulates CAR-T cell dysfunction and tumor antigen escape. J Signal Transduct Target Ther. (2023) 8:45. doi: 10.1038/s41392-023-01078-w
WebbMJSenerUVileRG. Current status and challenges of oncolytic virotherapy for the treatment of glioblastoma. Pharm (Basel). (2023) 16:793. doi: 10.3390/ph16060793
ChioccaEARabkinSD. Oncolytic viruses and their application to cancer immunotherapy. J Cancer Immunol Res. (2014) 2:295–300. doi: 10.1158/2326-6066.CIR-14-0015
OverdahlAAlexanderDTallóczyZSunQWeiYZhangWet al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. J Cell Host Microbe. (2007) 1:23–35. doi: 10.1016/j.chom.2006.12.001
HolmanHAMacLeanAR. Neurovirulent factor ICP34.5 uniquely expressed in the herpes simplex virus type 1 Delta gamma 134.5 mutant 1716. J Neurovirol. (2008) 14:28–40. doi: 10.1080/13550280701769999
GroomeirMLachmannSRosenfeldMRGutinPHWimmerE. Intergeneric poliovirus recombinants for the treatment of Malignant glioma. Proc Natl Acad Sci U.S.A. (2000) 97:6803–8. doi: 10.1073/pnas.97.12.6803
DesjardinsAGromeierMHerndonJE2ndBeaubierNBolognesiDPFriedmanAHet al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. (2018) 379:150–61. doi: 10.1056/NEJMoa1716435
XiongZRaphaelIOlinMOkadaHLiXKohanbashGet al. Glioblastoma vaccines: past, present, and opportunities. EBioMedicine. (2024) 100:104963. doi: 10.1016/j.ebiom.2023.104963
SchumacherTNSchreiberRD. Neoantigens in cancer immunotherapy. Science. (2015) 348:69–74. doi: 10.1126/science.aaa4971
LiJXiaoZWangDJiaLNieSZengXet al. The screening, identification, design and clinical application of tumor-specific neoantigens for TCR-T cell. Mol Cancer. (2023) 22:141. doi: 10.1186/s12943-023-01844-5
LinMJSvensson-ArvelundJLubitzGSMarabelleAMeleroIBrownBDet al. Cancer vaccines: the next immunotherapy frontier. Nat Cancer. (2022) 3:911–26. doi: 10.1038/s43018-022-00418-6
Cabeza-CabrerizoMCardosoAMinuttiCMPereira da CostaMReis e SousaC. Dendritic cells revisited. Annu Rev Immunol. (2021) 39:131–66. doi: 10.1146/annurev-immunol-061020-053707
YinXChenSEisenbarthSC. Dendritic cell regulation of T helper cells. Annu Rev Immunol. (2021) 39:759–90. doi: 10.1146/annurev-immunol-101819-025146
DatsiASorgRV. Dendritic cell vaccination of glioblastoma: road to success or dead end? Front Immunol. (2021) 12:770390. doi: 10.3389/fimmu.2021.770390
BotaDATaylorTHPiccioniDEDumaCMLaRoccaRVKesariSet al. Phase 2 study of AV-GBM-1 (a tumor-initiating cell targeted dendritic cell vaccine) in newly diagnosed Glioblastoma patients: safety and efficacy results. J Exp Clin Cancer Res. (2022) 41:344. doi: 10.1186/s13046-022-02552-6
CozziSNajafiMGomarMCiammellaPIottiCIaccarinoCet al. Delayed effect of dendritic cells vaccination on survival in glioblastoma: a systematic review and meta-analysis. Curr Oncol. (2022) 29:881–91. doi: 10.3390/curroncol29020075
LiauLMAshkanKBremSCampianJLTrusheimJEIwamotoFMet al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: a phase 3 prospective externally controlled cohort trial. JAMA Oncol. (2023) 9:112–21. doi: 10.1001/jamaoncol.2022.5374
Summary
Keywords
glioblastoma, innate immunity, tumor-associated macrophages, NK cell, microglia, myeloidderived suppressor cells, tumor microenvironment, immunotherapy
Citation
Zhang W, Zhang W, Wu H and Han X (2025) Harnessing innate immunity against glioblastoma microenvironment. Front. Immunol. 16:1648601. doi: 10.3389/fimmu.2025.1648601
Received
17 June 2025
Accepted
10 July 2025
Published
25 July 2025
Volume
16 – 2025
Edited by
Jin Bin, Shandong University, China
Reviewed by
Zhenghui Li, Zhengzhou University, China
Updates

Check for updates
Copyright
© 2025 Zhang, Zhang, Wu and Han.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinsheng Han, hansong2022@126.com
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.





