

Ethics statement
Ethical approval for the collection of serum residual samples from was provided by NHS Greater Glasgow and Clyde Biorepository (application 837).
Serum samples
Residual biochemistry serum samples were randomly collected from both primary care (general practice) and secondary care (hospital) settings by the NHSGGC Biorepository between March 31, 2020, and September 22, 2021. Informed consent for the use of serum samples was waived by the NHSGGC Biorepository, as the study used anonymised serum samples. Metadata including date of collection, sex, age, and vaccination history were provided, and the natural infection history of each patient was inferred from the date of last positive PCR test. Of the approximately 41,000 samples collected, a representative subsample of 350 sera was selected that maintained the underlying demographic proportions of the full sample by stratified random sampling. Metadata on the complete infection history of each individual was unavailable, and so categories of natural infection history likely include both singly- and multiply-exposed individuals.
Generation of lentiviral pseudotypes
Gene constructs containing the SARS-CoV (GenBank: AY394995.1), Rs4084 (GenBank: KY417144.1), GX/P1E (GISAID: EPI_ISL_410539), and RaTG13 (GenBank: MN996532.2) spike genes were codon-optimized and synthesized by GenScript Biotech. Each construct was cloned into an expression vector and co-transfected with p8.91-HIV (a gag-pol lentiviral core)60 and pCSFLW-firefly (which contains a luciferase reporter gene with HIV-1 packaging signal)61 into HEK293T cells using polyethylenimine. Cells were maintained at 37 °C, 5% CO2, in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2-mmol/l L-glutamine, 100 µg/ml streptomycin and 100IU/ml penicillin. Supernatants containing HIV (SARS-CoV-X) virions were harvested 48 h after transfection and frozen at −80 °C before use. All plasmids used in this study are available for reuse.
Neutralization assays
Pseudotype-based neutralization assays were performed as described previously62,63. Briefly, stably expressing HEK293-hACE2 (human angiotensin-converting enzyme 2) cells were generated by transfection of HEK293 cells with pSCRPSY-hACE2 and maintained as above. Serum samples were diluted 1:25 in Complete DMEM, and 25 µl of each diluted serum sample was transferred to the wells of a white 96-well plate. An equal volume (25 µl) of viral pseudotype was added to each well, resulting in a final serum concentration of 1:50. Plates were incubated for 1 h at 37 °C, 5% CO2, after which 50 µl of 4 × 105 cells/ml of HEK293-hACE2 cells were added to each well. Plates were then incubated for a further 48 h. After incubation, 100 µl of Steadylite Plus was added to each well, and the plate was incubated in the dark for 10 min. Luciferase activity in each well was quantified on a Perkin Elmer Ensight multimode plate reader. The proportion of pseudovirus neutralization was calculated as \({n}_{i}=1-\frac{{L}_{i}}{{L}_{c}}\), where \({L}_{i}\) is the luciferase activity of sample wells, and \({L}_{c}\) the luciferase activity of no-serum control wells. Two technical replicates for each combination of serum and pseudovirus were collected and averaged to provide final measures of neutralization. Where \({n}_{i} < 0\), which occurred when the luciferase activity of sample wells exceeded the activity of no-serum control wells, the proportion of neutralization was set to zero.
To estimate overall effects of vaccination and recovery from SARS-CoV-2 infection on neutralization, data from all serum groups were used to fit linear mixed effects models with a fixed effects structure \({y}_{i,r,v}={\beta }_{1,r}+{\beta }_{2,v}+{\beta }_{3,r,v}+e\). Here, \({y}_{i,r,v}\) is the percentage neutralization of serum from individual \(i\) with recovery status \(r\) and vaccine status \(v\). \({\beta }_{1,r}\), \({\beta }_{2,v}\), and \({\beta }_{3,r,v}\) represent the predicted contribution to percentage neutralization of recovery status, vaccination status, and their interaction, respectively, while \(e\) represents the model residuals. In this model, random effects of pseudotype virus identity and serum ID were included to account for between-group variability and repeated measures from individual sera. Differences in neutralization between viruses were estimated using a similar modelling approach but with virus identity included as a fixed effect and recovery and vaccine status as random effects. To estimate the overall effect of spike protein similarity on neutralization, data from all serum groups, excluding naïve + unvaccinated individuals, were used to fit a quadratic mixed effects model with a fixed effects structure \({y}_{i,s}={{\beta }_{s}}^{2}+e\). In this model, \({y}_{i,s}\) is the percentage neutralization of serum from individual \(i\) against spike protein of similarity \(s\); \({{\beta }_{s}}^{2}\) represents the predicted percentage neutralization against a spike protein of similarity \(s\). Random effects were included for recovery status, vaccination status, and serum ID. Similarity were calculated as the percentage amino acid identity to the Wuhan-Hu-1 spike protein sequence, taken from a translation alignment (described below).
Spike protein phylogeny
Spike protein coding sequences of SARS-CoV-2 (Wuhan-Hu-1 strain, GenBank: MN908947.3), SARS-CoV, Rs4084, GX/P1E, and RaTG13 (accessions in the previous section) were translated and aligned in MUSCLE64 with default settings. Phylogenetic inference was performed with BEAST version 10.5.065 using a 1 + 2 & 3 codon partition model with unique substitution rates and base frequencies for the partitions. Each partition was fitted to separate relaxed uncorrelated lognormal molecular clock models66 using random starting trees, four-category gamma-distributed HKY substitution models, and a birth-death process tree-shaped prior67. Four independent BEAST runs were performed, each with 100 million Markov Chain Monte Carlo (MCMC) iterations sampled every 1000 iterations. Runs were combined using LogCombiner with a burn-in of 20% and evaluated for convergence, sampling, and autocorrelation using Tracer version 1.7.268. A maximum clade credibility tree was inferred from the posterior sample and visualized with ggtree69.
Epidemiological model structure
An age-structured, stochastic SEIRS model was used to explore the population dynamics of co-circulating SARS-CoV-2 and SARS-CoV-X in the presence of vaccination (Fig. 2A). Within this structure, susceptible (S) individuals may encounter infectious individuals with SARS-CoV-2 (I2) or SARS-CoV-X (IX) and transition to a corresponding exposed compartment (E2 or EX), with the likelihood of S-to-E transition controlled by the transmission probabilities of each virus. Exposed individuals then transition to an infectious state (I2 or IX), with the probability of E-to-I transition inferred from the incubation rate of each virus, and from infectious to recovered (R2 or RX) with a probability inferred from the recovery rates of each virus. Recovered individuals are considered immune to the virus they have recovered from but may transition back to a naïve state over time, with the R-to-S transition probability calculated from the waning rates of infection-acquired immunity.
To allow for re-infection of recovered individuals by the circulating SARS coronavirus, recovered individuals may also, upon encountering an infectious individual, transition to a separate exposed state (E2RX or EXR2). These transitions occur with probabilities distinct from those of S-to-E, allowing immunity from one virus to reduce susceptibility to infection with the other. Re-infected individuals then transition through separate infectious compartments (I2RX or IXR2)—allowing re-infections to have distinct mortality and transition probabilities to infections of immunologically naïve individuals—before transitioning to the same recovered compartment (Rall) where individuals are refractory to both viruses. Unique EIR compartments exist for each SARS-CoV-2 variant (Wuhan, Alpha, Delta, Omicron), allowing their infection phenotypes to vary, and additional E and I compartments exist for individuals recovered from a previous SARS-CoV-2 variant that have been re-infected with a subsequent variant. This allows recovered individuals who are immune to one variant (e.g., Wuhan) to be re-infected with a later variant, albeit with some cross-immunity.
Three distinct levels of this base structure were used to represent different vaccination statuses (unvaccinated, “U”; protected by 1 dose, “VI”; and protected by 2 doses, “VII”). Individuals in a lower level of vaccine protection may transition to their equivalent compartment in a higher level of vaccine protection with a probability based on the current population vaccination rate, and from a higher level to unvaccinated at a probability based on the rate of decay of SARS-CoV-2 vaccine-derived antibodies. By representing vaccination in this way, a distinction is made between the current protection phenotype of an individual, which increases with vaccination and decreases with waning immunity, and an individual’s vaccination history, where the number of doses may only increase with time. This allows an individual with a vaccination history of many doses to have an unprotected phenotype, provided enough time has passed from their last dose for the protection to have waned. Given the timing of vaccinations during and after the COVID-19 pandemic in the model population (Scotland), a negligible number of individuals achieved a protected by 3 doses (“VIII”) phenotype throughout the model run, and so a fourth level representing these individuals was omitted for parsimony (Supplementary Fig. 2A). This model structure allows for individuals with immunity from both natural infection and vaccination (e.g., R2VII) to differ phenotypically from those with just natural (e.g., R2) and just vaccine (e.g., VII) sources of immunity, capturing some of the diversity in immune protection due to combinations of exposure and vaccine history.
Host population structure
The population of Scotland was used as the basis for the host model population, for which there is detailed demographic and epidemiological information from before the COVID-19 pandemic to the present. Where information directly describing this population was unavailable, such as in the case of social contact data during and after the pandemic, information from the UK-wide population was used. Each compartment of the model was sub-divided into 16 age groups of 5 years, beginning 0-4 years old and ending 75+ years old, and the starting population sizes of each group were set to the projected population estimates from the National Records of Scotland for 2020. The birth rates of each age group were set as 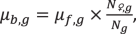 , where \({\mu }_{b,{g}}\) is the birth rate, \({\mu }_{f,{g}}\) the fertility rate, N♀,g the female population size, and \({N}_{g}\) the total population size of age group \(g\). All births were added to the 0-4 age group, and increases in age group occurred deterministically at a daily rate of \(\frac{1}{365\times 5}\). Age-stratified mortality and net migration rates were taken from publicly available data (see Supplementary Table 7 for all parameterisation sources). Migrants were assumed to have the same epidemiological properties as the Scotland population at the point of migration. For example, if 5% of the model population were in the I2 compartment, each migrant entering the model at that time point had a probability of 0.05 of being infectious for SARS-CoV-2. These parameters together created a host population that was largely stable over the 8-year duration of the model runs, with size and age progression similar to official population projections (Supplementary Fig. 10).
, where \({\mu }_{b,{g}}\) is the birth rate, \({\mu }_{f,{g}}\) the fertility rate, N♀,g the female population size, and \({N}_{g}\) the total population size of age group \(g\). All births were added to the 0-4 age group, and increases in age group occurred deterministically at a daily rate of \(\frac{1}{365\times 5}\). Age-stratified mortality and net migration rates were taken from publicly available data (see Supplementary Table 7 for all parameterisation sources). Migrants were assumed to have the same epidemiological properties as the Scotland population at the point of migration. For example, if 5% of the model population were in the I2 compartment, each migrant entering the model at that time point had a probability of 0.05 of being infectious for SARS-CoV-2. These parameters together created a host population that was largely stable over the 8-year duration of the model runs, with size and age progression similar to official population projections (Supplementary Fig. 10).
To represent changes in social contacts over time, questionnaire data from multiple social mixing studies were used to calculate age-specific per-person per-day contact rates for different time periods. For the pre-lockdown period of 01/01/2020 (dd/mm/yyyy) to 23/03/2020, rates were set to those of the 2008 POLYMOD study of the UK population70, with the study’s contact sub-categories (“home”, “work”, “school”, and “other”) summed to produce an overall daily rate of contact. For the period of 24/03/2020 to 02/03/2022, UK social contact information was collected continuously by the CoMix study71. In this study, the authors split their data into nine timeframes describing different lockdown and easing periods. To provide a finer temporal scale to the epidemiological model, this data was re-analysed to provide weekly estimates of contact rate using the same approach as the original study. Briefly, participant contacts were censored to a daily maximum of 50, and the mean number of contacts inferred from a negative binomial model of structure \({m}_{i,j}={\beta }_{1,i}+{\beta }_{2,j}+{\beta }_{3,i,j}+e\). Here, \({m}_{i,j}\) represents the mean number of contacts of participant age group \(i\) to contact age group \(j\); \({\beta }_{1,i}\) is the effect of participant group \(i\) on the mean number of contacts; \({\beta }_{2,j}\) is the effect of contact group \(j\) on the mean number of contacts; \({\beta }_{3,i,j}\) is the interaction effect of participant group \(i\) and contact group \(j\) on the mean number of contacts; and \(e\) is the model residuals.
Symmetrical per-capita contact matrices were calculated from the output of this model using the following equation: \({m{\prime} }_{i,j}=\frac{1}{{2N}_{i}}\left({m}_{i,j}{N}_{i}+{m}_{j,i}{N}_{j}\right)\). Here, \({m{\prime} }_{i,j}\) is the adjusted mean number of contacts, \({N}_{i}\) the total number of individuals in age group \(i\), and \({N}_{j}\) the total number of individuals in age group \(j\). A follow-up to the CoMix study was conducted with data collected from 17/11/2022 to 07/12/2022, providing the most up-to-date estimates of UK contact rates available72. Data from this study was used to set the model contact rates from 17/11/2022 to the end of the model run (01/01/2028). Weekly contact rates in the inter-study period of 03/03/2022 to 16/11/2022 were inferred by linear interpolation from the final week of data of the original study to the first week of the follow-up study.
Virus and immune phenotype parameterization
Parameter values for the infection-related mortality rates and incubation rates (E-to-I) of SARS-CoV and each SARS-CoV-2 variant were obtained from the literature (Supplementary Table 7). Literature estimates of the recovery rate (I-to-R), naturally-acquired immunity waning rates (R2-to-S2), and \({R}_{0}\) for SARS-CoV-2 were highly variable and showed little consensus. Instead, these parameters were calibrated by fitting the epidemiological model to prevalence data taken from the UK Coronavirus Infection Survey30, using an approximate Bayesian computation (ABC) approach as follows.
Log-normal priors were used for the \({R}_{0}\) of each variant (µ = 1, σ = 0.5 on the log scale), corresponding to a median \({R}_{0}\) of ~3.0 and a 95% prior credible interval (CI) of ~1–7.2. Logit-normal priors were used for the recovery rates (µ = −1, σ = 0.6 on the logit scale), corresponding to a median of 0.26 and a 95% prior CI of 0.10–0.54, and for the immune waning rates (µ = −5, σ = 2 on the logit scale), corresponding to a median of 0.0067 and a 95% prior CI of 0.0001–0.25. Due to the sequential non-independence of parameters (the values of subsequent variants are highly-dependent on the calibrated estimates of previous variants), calibration was performed separately for each variant in order of emergence. For each variant, 2.5 million randomly sampled parameter sets were run for 10 replicate model iterations, and mean model-estimated SARS-CoV-2 prevalence was calculated for each timepoint present in the Coronavirus Infection Survey. Parameter sets were filtered to remove simulations with a RMSE > 0.01, corresponding to an average deviation of > 1% from the observed SARS-CoV-2 prevalence. For the remaining parameter sets, Epanechnikov kernel weights were applied to scaled RMSE values to construct a smooth approximation of the posterior distribution. To propagate uncertainty in these parameter values forward, all subsequent models included in this study sampled unique values from each approximate posterior for each independent model iteration. For SARS-CoV, literature estimates of \({R}_{0}\) were available, but no estimates were available for the recovery rate. Additionally, no information on the infection phenotypes of the prospective zoonotic SARS coronaviruses in humans exists. In the absence of this information, all missing phenotypes were assumed to equal those of the most closely related SARS coronavirus with a known phenotype. For example, the \({R}_{0}\) of Rs4084 was set equal to SARS-CoV, while those of RaTG13 and GX/P1E were set equal to the SARS-CoV-2 Wuhan variant.
Transmission rates were calculated from the following \({R}_{0}\) equation, derived from the SEIRS model structure: \({R}_{0,v}=\frac{{\varepsilon }_{v}{\beta }_{v}{m}^{{\prime} }}{\left({\varepsilon }_{v}+\lambda \right)\left({\gamma }_{v}+{\delta }_{v}+\lambda \right)}\). Here, \({\beta }_{v}\) is the transmission rate; \({R}_{0,v}\) the basic reproductive number; \({\varepsilon }_{v}\) the incubation rate; \({\gamma }_{v}\) the recovery rate; and \({\delta }_{v}\) the infection-related mortality rate of virus \(v\). \(\lambda\) is the population crude death rate and \(m{\prime}\) the mean per-day per-capita contact rate. The transmission rates and infection-related mortality rates above describe the phenotypes of infection in naïve and unvaccinated individuals. Estimates of the proportion with which infection-related mortality is reduced in recovered and vaccinated individuals were taken from the literature. The proportion with which transmission rates are reduced in recovered and vaccinated individuals for SARS-CoV-X infections is assumed to correspond linearly (\(1-x\)) to the pseudotype neutralization data in Fig. 1, and for SARS-CoV-2 re-infections from62 (Supplementary Fig. 9). The exact relationship between neutralization titres and protection from sarbecovirus infection is unknown, with both linear and non-linear relationships reported in the literature5.
The waning rates of immunity in recovered individuals (R-to-S) and vaccinated individuals (VI-to-S and VII-to-S) were calculated separately from the half-lives of infection and vaccine derived antibodies against SARS-CoV-2 with the following exponential decay equation: \({\mu }_{w,v}=\frac{-{{\mathrm{ln}}}(0.5)}{{t}_{\frac{1}{2},i}}\). Here, \({\mu }_{w,v}\) represents the waning rate, and \({t}_{\frac{1}{2},v}\) the antibody half-life of immunity of type \(i\) (natural or vaccine).
Age-stratified phenotypes
There is strong evidence in the literature that immune protection and infection-related mortality for coronavirus infections vary across age groups. To represent this variation, non-linear least squares models, fitted using the nls function from the R core stats package73, were used to infer how the relationship between phenotype and age changes with the population parameter value. For infection-related mortality, age-stratified data on SARS-CoV-2 and MERS-CoV were used to fit a four-parameter logistic function (Supplementary Fig. 11) as \({\delta }_{v,a}={L}_{2}+{\beta }_{1}{\delta }_{v}+\frac{{\beta }_{2}{\delta }_{v}+{L}_{1}-{L}_{2}}{1+{e}^{-(k+{\beta }_{3{\delta }_{v}})(a-{a}_{0}+{\beta }_{4}{\delta }_{v})}}\),. Here, \({\delta }_{v,a}\) is the infection-related mortality rate of virus \(v\) for hosts of age \(a\); \(k\) is the slope, \({L}_{1}\) the upper plateau, and \({L}_{2}\) the lower plateau of the logistic function; and \({a}_{0}\) is the inflection point. \({\beta }_{1}{\delta }_{v}\), \({\beta }_{2}{\delta }_{v}\), \({\beta }_{3}{\delta }_{v}\), and \({\beta }_{4}{\delta }_{v}\) describe how the population infection-related mortality rate affects the lower plateau, upper plateau, slope, and midpoint of the logistic function, respectively.
Evidence for age-stratified effects of vaccine-efficacy are also prevalent in the literature, although effect sizes are typically minor74,75,76,77,78,79. The effectiveness of the second dose of the COVID-19 vaccine, administered at different intervals from the first dose, was used to provide a dataset where the population-level estimate of immune protection varied, and the age-stratified estimates of immune protection were known (Supplementary Fig. 12). This data was used to fit a model with a formula composed of two three-parameter logistic functions (one describing the increase in immune effectiveness from birth to middle age, the other describing the decline in effectiveness from middle age to old age), and an intercept influenced by the population-level protection: \({p}_{i,a,v}={L}_{3}+{\beta }_{1}{p}_{i,v}+\frac{{L}_{1}}{1+{e}^{-{k}_{1}(a-{a}_{{{\mathrm{1,0}}}})}}+\frac{{L}_{2}}{1+{e}^{-{k}_{2}(a-{a}_{{{\mathrm{2,0}}}})}}\). Here, \({p}_{i,a}\) is the level of protection conferred by immunity source \(i\) against virus \(v\) on age group \(a\). \({L}_{1}\), \({k}_{1}\), and \({a}_{1,o}\) are the upper plateau, slope, and infection point of the increase in immune effectiveness with age, while \({L}_{2}\), \({k}_{2}\), and \({a}_{2,o}\) are the equivalents for the decrease in immune effectiveness with age. \({\beta }_{1}{p}_{i,v}\) describes the effect of population-level immune protection on the function’s intercept (\({L}_{3}\)). Little consensus exists for an effect of host age on the incubation or recovery rates of SARS coronavirus infections80,81,82,83,84, and so these parameters are assumed to be equal across age groups.
Vaccination rates
For the initial COVID-19 vaccination program (08/12/2020 to 11/09/2022), daily vaccination rates for each age group receiving the first (\({u}_{{D}_{1}}\)), second (\({u}_{{D}_{2}}\)), and third (\({u}_{{D}_{3}}\)) vaccine doses were calculated from publicly available data85. As vaccination exists in the epidemiological model as a transient phenotype with three levels (U, VI, VII), two distinct rates exist: the transition rate from naïve to protected with 1 dose (\({u}_{U\to {V}_{I}}\)) and the transition rate from protected with 1 dose to protected with 2 doses (\({u}_{{V}_{I}\to {V}_{II}}\)). As entry into the VII level is impossible while only 1 dose of the vaccine has been administered, this rate was set as \({u}_{{V}_{I}\to {V}_{II}}={u}_{{D}_{2}}+{u}_{{D}_{3}}\). Transitioning from a naïve to vaccinated phenotype can be produced at any point in the vaccination program and by any dose in a patient’s vaccination history, and so this rate was set as \({u}_{U\to {V}_{I}}={u}_{{D}_{1}}+{u}_{{D}_{2}}+{u}_{{D}_{3}}\). Vaccination rates by age group during the booster programs in Winter 2022, Spring 2023, Winter 2023, and Spring 2024 were used to extend \({u}_{{D}_{3}}\) to 14/07/202486. Spring and Winter booster programs were then assumed to recur annually at rates identical to Winter 2023 and Spring 2024 for the remainder of the model.
The rate of administration of the first COVID-19 vaccine dose over time in Scotland is well approximated by a non-normalised Gaussian curve (Supplementary Fig. 13), and so a similar curve with a mean (µ) of 30 days and standard deviation (σ) of 10 days was used to define a theoretical preventative campaign against SARS-CoV-X emergence lasting 60 days: \(r\left(t\right)=A{e}^{-\frac{{(t-\mu )}^{2}}{{2\sigma }^{2}}}\). By adjusting the scaling factor (A), the vaccination rates described by this curve result in different cumulative vaccine uptake levels over the course of the campaign. Solutions for A that produce specific values of vaccine uptake were calculated using root-finding methods with the uniroot function in R73.
Stochasticity
Mean rates for all model parameters (excluding the aging rate) were converted to probabilities using a Euler scheme with binomial increments and exponential rates87 such that the number of individuals \(N\) transitioning from compartment \(A\) to \(B\) at time \(t\) is given as \({N}_{A\to B,t}={Binomial}({N}_{A,t-1},{1-e}^{{-\mu }_{A\to B,t}})\). Here, \({Binomial}(n,{p})\) represents the binomial distribution, \(n\) is the number of individuals available to a given transition, and \(p\) is the probability of transition calculated from the mean rate \(u\).
Model run conditions
The model was implemented and run using the Odin88 and Odin Dust89 R packages, with random starting seeds, a 1-day time step, and a duration of 8 years spanning 1st January 2020 to 1st January 2028. To introduce the SARS-CoV-2 Wuhan variant, five exposed individuals (E2Wuhan) were added to the model on 23rd February 2020. This date was inferred from the description of the first confirmed positive case of SARS-CoV-2 infection in Scotland90, which was reported on 2nd March 2020 in an individual presenting 3 days after symptom onset (-3 days) and after a ~ 5-day incubation period (-5 days). Five individuals were chosen as this ensured SARS-CoV-2 reached endemicity in all model runs conducted in this study. Introduction of new SARS-CoV-2 variants to the model occurred deterministically by taking the total number of SARS-CoV-2 infectious individuals, regardless of variant, and multiplying by the prevalence of each variant as reported for the Scotland population over time. These values were then used in conjunction with contact probabilities to calculate the number of individuals exposed to each variant as the model progressed. A single 30-year-old SARS-CoV-X individual (EX) was introduced to the model on 23rd February 2024, four years after the initial SARS-CoV-2 exposure.
Sensitivity analysis
To provide a sufficient sampling of parameter space to allow for a Sobol sensitivity analysis on nine parameters—the \({R}_{0}\) of SARS-CoV-2 and SARS-CoV-X; the levels of natural and vaccine-acquired cross-immune protection against SARS-CoV-X; the timing and uptake of the preventative vaccination program; and the waning rates of vaccine-acquired immunity, naturally-acquired SARS-CoV-2 immunity, and naturally-acquired SARS-CoV-X immunity—a machine learning emulator was trained on the output of the epidemiological model as follows. A Sobol sample (n = 500,000) of nine-dimensional parameter space was produced using uniform distributions of each parameter (Table 1). Each parameter set in the Sobol sample scheme was run for a small number of iterations (500), providing a dense but high-Monte-Carlo-noise exploration of parameter space, an approach that has been shown to efficiently provide training data for model emulation91. The probability of SARS-CoV-X emergence (emergences/trials) was then used to train a gradient-boosted regression trees (GBRT) model using the R package XGBoost92, configured with a maximum tree depth of 9 (equal to the number of explanatory variables), a learning rate of 0.01, a subsampling fraction of 0.7, and all available predictors included at each tree split. Ten-fold cross-validation was used within XGBoost to identify the optimal number of boosting iterations to avoid overfitting the training data. An additional 5000 parameter sets were produced using a Latin Hypercube sample of parameter space with the same uniform distributions on each parameter. Each of these parameter sets were run for 5000 model iterations to produce a low-density, low-Monte-Carlo-noise test dataset. When evaluated on the independent test dataset, the trained emulator closely reproduced the epidemiological model outputs, achieving an R2 > 0.997 with errors consistent across predicted values (Supplementary Fig. 14). Sensitivity analysis was then conducted using variance-based Sobol indices implemented in the R package Sensitivity93. A Saltelli paired-sample design was generated from two \({{\mathrm{200,000}}}\times 9\) Sobol sampling matrices, for a total of 2.2 million parameter sets. Predicted probabilities of SARS-CoV-X emergence for each parameter set were taken from the GBRT emulator and used to calculate first-order and total-order Sobol indices, with 1000 bootstrap replicates to estimate confidence intervals.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.




