

Mutations of LET7 suppress mekk1–mkk1
mkk2–mpk4 autoimmunity
Silencing MEKK1 via Agrobacterium-mediated VIGS in Arabidopsis wild-type (WT) Columbia (Col-0) plants triggered plant growth defects and dwarfism, which were alleviated in the let7-1 mutant (salk_021153) (Fig. 1a). Notably, let7-1 did not suppress RNAi-BAK1/SERK4-triggered growth defects (Fig. 1a), suggesting the specificity of let7-1 suppression of autoimmunity. RNAi-MEKK1-triggered cell death, as evidenced by trypan blue staining, and elevated H2O2 accumulation detected by 3,3′-diaminobenzidine (DAB) staining were also suppressed in let7-1 (Fig. 1b). Moreover, elevated expression of PATHOGENESIS-RELATED 1 (PR1), PR2 and PR5 triggered by RNAi-MEKK1 was compromised in let7-1 (Fig. 1c).
Fig. 1: The let7 mutants suppress mekk1-triggered autoimmunity.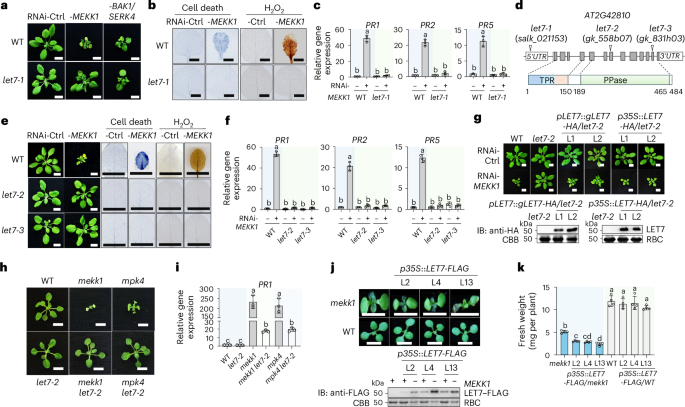
a, The let7-1 mutant suppresses growth defects triggered by RNAi-MEKK1 but not RNAi-BAK1/SERK4. WT and let7-1 plants are shown three weeks after inoculation with Agrobacterium tumefaciens carrying the VIGS vector targeting GFP as a control (Ctrl), RNAi-MEKK1 or RNAi-BAK1/SERK4. Scale bars, 1 cm. b, The let7-1 mutant suppresses cell death and H2O2 production triggered by RNAi-MEKK1. Detached true leaves after VIGS from a were stained with trypan blue for cell death or DAB for H2O2 accumulation. Scale bars, 0.5 cm. c, The let7-1 mutant suppresses PR1, PR2 and PR5 expression triggered by RNAi-MEKK1. RT-qPCR was performed with samples from a. The expression levels of PR1, PR2 and PR5 were normalized to ACTIN2. PR1: Column 1 versus Column 2, P < 0.0001, Column 3 versus Column 4, P = 0.9749; PR2: Column 1 versus Column 2, P < 0.0001, Column 3 versus Column 4, P = 0.661; PR5: Column 1 versus Column 2, P < 0.0001, Column 3 versus Column 4, P = 0.1462. d, Schematic diagram of mutant lines and protein motifs of LET7. Top: the LET7 (AT2G42810) gene is shown with three T-DNA insertional lines. The white boxes indicate 5′ and 3′ untranslated regions (UTRs) of mRNA, the grey boxes indicate exon protein-coding regions and the lines indicate introns. Bottom: protein motifs, including the TPR domain and the PPase catalytic domain, with amino acid positions labelled. e, The let7-2 and let7-3 mutants suppress growth defects, cell death and H2O2 production triggered by RNAi-MEKK1. The experiments were performed similarly to those in a and b. Scale bars, 1 cm. f, The let7-2 and let7-3 mutants suppress PR1, PR2 and PR5 induction triggered by RNAi-MEKK1. The experiments were performed similarly to those in c. PR1: Column 1 versus Column 2, P < 0.0001, Column 3 versus Column 4, P = 0.6921, Column 5 versus Column 6, P = 0.5459; PR2: Column 1 versus Column 2, P < 0.0001, Column 3 versus Column 4, P = 0.87, Column 5 versus Column 6, P = 0.6896; PR5: Column 1 versus Column 2, P < 0.0001, Column 3 versus Column 4, P > 0.9999, Column 5 versus Column 6, P = 0.7966. g, Complementation of let7-2 with LET7 restores the growth defects triggered by RNAi-MEKK1. The genomic DNA of LET7 with the HA tag under its native promoter (pLET7::gLET7-HA) or cDNA of LET7 with the HA tag under the CaMV 35S promoter (p35S::LET7-HA) was transformed into let7-2. VIGS was performed similarly to that in a. Two independent homozygous complementation lines for each construct are shown. Scale bars, 1 cm. Bottom: immunoblots (IBs) using an anti-HA antibody show LET7–HA proteins, and Coomassie brilliant blue (CBB) staining of Rubisco proteins (RBC) serves as a loading control. The molecular weight (kDa) is labelled on the left of the regular SDS–PAGEs for all immunoblots in this study. h, The let7-2 mutant suppresses the growth defects of the mekk1 and mpk4 mutants. Three-week-old plants grown on half-strength Murashige and Skoog (1/2 MS) plates with the indicated genotypes are shown. Scale bars, 0.5 cm. i, The let7-2 mutant suppresses the elevated expression of PR1 in mekk1 and mpk4. The expression of PR1 was normalized to that of ACTIN2. Column 1 versus Column 2, P > 0.9999, Column 1 versus Column 3, P < 0.0001, Column 3 versus Column 4, P < 0.0001, Column 1 versus Column 5, P < 0.0001, Column 5 versus Column 6, P < 0.0001. j,k, Overexpression of LET7 aggravates the growth defects of mekk1. The cDNA of LET7 with the FLAG tag under the 35S promoter (p35S::LET7-FLAG) was transformed into the heterozygous mekk1 mutant (MEKK1+/−). Three independent lines (L2, L4 and L13) were genotyped to identify mekk1 homozygous mutants (mekk1 or −MEKK1) and WT (WT or +MEKK1) plants. Seven-day-old representative plants are shown (j, top). Scale bars, 0.5 cm. LET7–FLAG proteins in transgenic plants are shown by immunoblotting using an anti-FLAG antibody with CBB-stained RBC as loading controls (j, bottom). The fresh weights were quantified (k). Column 1 versus Column 2, P = 0.0096, Column 1 versus Column 3, P = 0.0012, Column 1 versus Column 4, P = 0.0266, Column 1 versus Column 5, P < 0.0001, Column 5 versus Column 6, P = 0.9685, Column 5 versus Column 7, P = 0.9961, Column 5 versus Column 8, P = 0.225. The data are shown as mean ± s.d. (n = 3 biologically independent samples in c, f and i, and n = 4 biologically independent plants in k). Different letters (c, f, i and k) indicate significant differences determined by one-way analysis of variance followed by Tukey’s test (P < 0.05). The experiments were repeated at least four times in a–c, e, f, h and i and three times in g, j and k with similar results.
The let7-1 mutant was annotated to carry a T-DNA insertion in the 5′ untranslated region of AT2G42810, which encodes a TPR-domain-containing protein phosphatase (Fig. 1d). To validate the role of LET7 in controlling MEKK1-mediated cell death, we analysed two additional alleles, let7-2 (gk_558b07) and let7-3 (gk_831h03), with reduced LET7 expression compared with WT plants (Fig. 1d and Extended Data Fig. 1a). Similar to let7-1, both let7-2 and let7-3 mutants suppressed RNAi-MEKK1-triggered growth defects, cell death and H2O2 accumulation (Fig. 1e), as well as the elevated expression of PR1, PR2 and PR5 (Fig. 1f). In addition, expressing the genomic DNA of LET7 (gLET7) tagged with a double hemagglutinin (HA) epitope at the carboxy terminus under its native promoter (pLET7::gLET7-HA) in let7-2 restored RNAi-MEKK1-induced growth defects (Fig. 1g), cell death (Extended Data Fig. 1b), H2O2 accumulation (Extended Data Fig. 1c) and upregulation of PR1 (Extended Data Fig. 1d). Furthermore, overexpression of the LET7 coding region tagged with HA under the constitutive 35S promoter (p35S::LET7-HA) also restored RNAi-MEKK1-triggered growth defects (Fig. 1g). These data support the idea that LET7 plays an important role in regulating RNAi-MEKK1-triggered cell death.
MAPKK MKK1/2 and MAPK MPK4 function downstream of MEKK1 in SUMM2-mediated cell death. Similar to mekk1, the mkk1 mkk2 and mpk4 mutants are seedling lethal10. To delineate the genetic position of let7 in suppressing mekk1–mkk1 mkk2–mpk4 cell death, we generated the mekk1 let7-2 and mpk4 let7-2 double mutants via genetic crosses (Extended Data Fig. 1e,f). The mekk1 let7-2 and mpk4 let7-2 mutants suppressed the seedling lethality (Fig. 1h) and PR1 and PR2 accumulation of mekk1 and mpk4 (Fig. 1i and Extended Data Fig. 1g). Additionally, overexpression of LET7 tagged with a FLAG epitope under the 35S promoter (p35S::LET7-FLAG) aggravated the growth defects of mekk1 (Fig. 1j,k). Taken together, these data support the idea that LET7 functions genetically downstream of the MEKK1–MKK1/2–MPK4 cascade in SUMM2-mediated cell death.
LET7 phosphatase activity is necessary and sufficient to trigger autoimmunity
LET7 encodes a conserved PP5 across plant and animal kingdoms, characterized by a TPR domain and a catalytic PPase domain28,29 (Fig. 2a). Arabidopsis has one PP5 phosphatase, originally designated as phytochrome-associated protein phosphatase 5 (PAPP5)30 or AtPP5 (ref. 31). To reflect its role in regulating mekk1 cell death and align with the standardized nomenclature for Arabidopsis genes using a three-letter symbol (https://www.arabidopsis.org/portals/overview), the gene name LET7 was retained. To unravel which domain of LET7 regulates cell death, we generated transgenic plants carrying the LET7TPR or LET7PPase truncation with the HA tag under the 35S promoter (p35S::LET7TPR-HA or p35S::LET7PPase-HA) in let7-2. Expressing LET7TPR failed to complement RNAi-MEKK1-triggered growth defects in let7-2 (Fig. 2b). In contrast, transgenic plants with moderate expression of LET7PPase partially restored RNAi-MEKK1-triggered growth defects in let7-2 (Fig. 2c). These data imply that LET7PPase is sufficient to trigger cell death.
Fig. 2: The PPase domain with phosphatase activity is essential and sufficient to trigger cell death.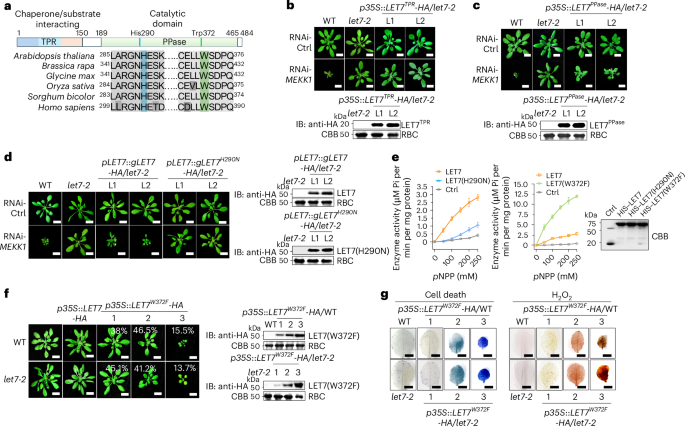
a, The conserved PPase domain with invariant histidine and tryptophan residues of the PP5 family in Arabidopsis thaliana, Brassica rapa, Glycine max, Oryza sativa, Sorghum bicolor and Homo sapiens. b, Overexpression of the TPR domain of LET7 (LET7TPR) cannot restore RNAi-MEKK1-triggered cell death in let7-2. L1 and L2 are two independent homozygous lines carrying p35S::LET7TPR-HA in let7-2. VIGS was performed similarly to that in Fig. 1a. Scale bars, 1 cm. The immunoblot with an anti-HA antibody shows LET7TPR–HA proteins (top) with CBB staining of RBC as a loading control (bottom). c, Overexpression of the PPase domain of LET7 (LET7PPase) partially restores RNAi-MEKK1-triggered cell death in let7-2. L1 and L2 are two independent homozygous lines carrying p35S::LET7PPase-HA in let7-2. Scale bars, 1 cm. d, Unlike LET7, expression of LET7(H290N) in let7-2 cannot restore the growth defects triggered by RNAi-MEKK1. Left: independent homozygous lines carrying pLET7::gLET7-HA (L1 and L2) or pLET7::gLET7H290N-HA (L1 and L2) in let7-2 were subjected to VIGS. Scale bars, 1 cm. Right: immunoblots with an anti-HA antibody show LET7–HA and LET7(H290N)–HA proteins with CBB staining of RBC as a loading control. e, Phosphatase activity is much reduced in LET7(H290N) and elevated in LET7(W372F) compared with LET7. An equal amount of LET7, LET7(H290N) or LET7(W372F) proteins purified from E. coli was incubated with a series of concentrations of pNPP as substrates in the phosphatase buffer. HIS–SUMO served as a control (Ctrl). The turnover of pNPP was measured photometrically by recording the absorption at 405 nm, and enzyme activity was calculated from the Beer–Lambert law. The data are shown as mean ± s.d. (n = 3, biologically independent samples). CBB staining of proteins is shown on the right. f,g, Overexpression of LET7(W372F) induces growth defects, cell death and H2O2 production. Unlike transgenic plants carrying p35S::LET7-HA, transgenic plants carrying p35S::LET7W372F-HA in the WT or let7-2 background show growth defects (f). Four-week-old soil-grown plants are shown with the percentage of plants displaying the indicated phenotype among a total of 174 transgenic plants for p35S::LET7W372F-HA WT and 182 transgenic plants for p35S::LET7W372F-HA let7-2 (f, left). Scale bars, 1 cm. Immunoblots with an anti-HA antibody show the protein expression of LET7(W372F)–HA (f, right). The true leaves of transgenic plants were used for trypan blue staining for cell death (g, left) and DAB staining for H2O2 production (g, right). Scale bars, 0.5 cm. The experiments were repeated four times in b–d and three times in e–g with similar results.
The LET7 His290 residue, corresponding to human HsPP5 His304, resides in the catalytic core, potentially for phosphate binding, and His290 substitution by asparagine blocked its PPase activity25,31,32 (Fig. 2a). Transgenic plants expressing LET7(H290N) under the native or 35S promoter no longer restored RNAi-MEKK1-triggered growth defects (Fig. 2d and Extended Data Fig. 2a,b) or PR1 expression (Extended Data Fig. 2c) in let7-2. Additionally, LET7(H290N) purified from Escherichia coli reduced the PPase activity in dephosphorylating p-nitrophenyl phosphate (pNPP) in an in vitro phosphatase activity, compared with LET7 (Fig. 2e). Furthermore, LET7(W372F) carrying the tryptophan-to-phenylalanine substitution W372F and resembling the HsPP5 hyperactive mutant W386F in the catalytic cleft33, exhibited a markedly enhanced PPase activity compared with LET7 (Fig. 2e). Transgenic plants overexpressing LET7W372F, but not LET7, under the 35S promoter (p35S::LET7W372F-HA) in the WT or let7-2 showed growth defects correlated with LET7(W372F)–HA protein levels (Fig. 2f and Extended Data Fig. 2d), cell death and H2O2 accumulation (Fig. 2g), and elevated PR1 and PR2 levels (Extended Data Fig. 2e). These findings indicate that the protein phosphatase activity of LET7 is positively correlated with autoimmunity.
LET7TPR interacts with and suppresses LET7PPase phosphatase activity and cell death inducibility
LET7PPase also triggered cell death in Nicotiana benthamiana (Fig. 3a). Additionally, Arabidopsis transgenic plants carrying p35S::LET7PPase-HA in the WT showed variable degrees of growth defects, with the severity correlated with LET7PPase–HA protein levels (Fig. 3b). These data imply that LET7PPase alone can trigger autoimmunity. CRCK3 and SUMM2 function genetically downstream of the MEKK1–MKK1/2–MPK4 cascade in cell death control9,15,20. LET7PPase–HA-triggered plant growth defects were partially alleviated in crck3 and summ2 mutants (Fig. 3b), suggesting that CRCK3 and SUMM2 are required for LET7PPase-triggered autoimmunity.
Fig. 3: LET7TPR interacts with LET7PPase and inhibits its phosphatase activity.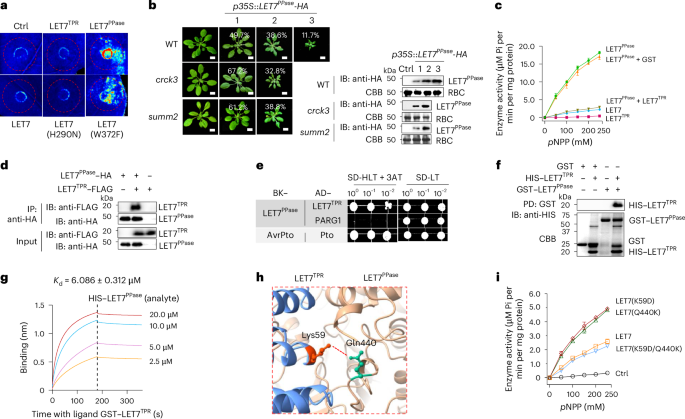
a, Unlike the full-length LET7 or LET7TPR, LET7PPase triggers cell death in N. benthamiana. A vector control (Ctrl), LET7, LET7TPR, LET7PPase, LET7(H290N) or LET7(W372F) was expressed in N. benthamiana leaves via agroinfiltration, and cell death in infiltrated areas is shown at two days post-infiltration under UV light. b, Overexpression of LET7PPase in Arabidopsis triggers cell death, which is alleviated in crck3 and summ2-8 mutants. LET7PPase with an HA tag under the 35S promoter (p35S::LET7PPase-HA) was transformed into WT (127 plants), crck3 (114 plants) or summ2 (108 plants). The transgenic plants were grouped into three categories (1, 2 and 3) on the basis of the severity of plant dwarfisms, with the percentages of each category shown in the figure. Scale bars, 1 cm. The protein expression of LET7PPase–HA is shown via immunoblotting using an anti-HA antibody with CBB-stained RBC as a loading control. c, LET7TPR suppresses the phosphatase activities of LET7PPase. The experiments were performed similarly to those in Fig. 2e. The same amount of GST proteins was added as a control for LET7TPR. d, LET7PPase associates with LET7TPR in Co-IP assays. LET7PPase–HA was co-expressed with LET7TPR–FLAG or vector control (−) in Arabidopsis protoplasts. Total proteins were immunoprecipitated with anti-HA affinity beads, followed by immunoblotting using an anti-FLAG or anti-HA antibody (top two panels). Proteins before immunoprecipitation were immunoblotted and are shown as input controls (bottom two panels). e, LET7PPase interacts with LET7TPR in Y2H assays. Yeast was grown on a synthetic drop-out (SD) medium without leucine and tryptophan (SD-LT) or without leucine, tryptophan and histidine (SD-HLT) supplemented with 1 mM 3-amino-1,2,4-triazol (3-AT). pGADT7 (AD) and pGBKT7 (BK) are vectors with AvrPto, Pto and PARG1 as controls. f, LET7PPase interacts with LET7TPR in in vitro pull-down (PD) assays. GST or GST–LET7PPase immobilized on glutathione agarose was incubated with HIS–LET7TPR proteins. Top: washed beads were pelleted for immunoblotting using an anti-HIS antibody. Bottom: input proteins were stained with CBB before pull-down. g, LET7PPase interacts with LET7TPR in BLI binding assays. Recombinant GST–LET7TPR proteins purified from E. coli were immobilized on GST biosensor chips and incubated over a range of concentrations (2.5–20 μM) of soluble HIS–LET7PPase proteins as the analyte. The plot shows the association (0 to 180 s) and dissociation (180 to 360 s) times of the interaction. The equilibrium Kd of LET7PPase and LET7TPR binding is 6.086 ± 0.312 μM as determined by Octet BLI analysis software. h, A close-up view of a salt bridge between Lys59 and Gln440 in the interface of the LET7PPase and LET7TPR domains. This structure was predicted by AlphaFold2. Lys59 and Gln440 are marked in orange and green, respectively. The interacting residues are in the stick representation. The dashed line indicates a salt bridge. i, Disruption of the salt bridge between Lys59 and Gln440 partially releases LET7 autoinhibition. Equal amounts of LET7, LET7(K59D), LET7(Q440K) or LET7 (K59D/Q440K) proteins were subjected to phosphatase assays. The experiments were repeated four times in a and f and three times in b–e and g–i with similar results. The data are shown as mean ± s.d. (n = 3 biologically independent samples in c and i).
In contrast to LET7PPase, neither LET7 nor LET7TPR elicited cell death in N. benthamiana (Fig. 3a), suggesting that LET7TPR may suppress LET7PPase-triggered cell death, in line with the autoinhibition of PPase activities by the TPR domain24,25. Indeed, LET7TPR inhibited the LET7PPase phosphatase activity, with LET7PPase showing a much stronger phosphatase activity than LET7 (Fig. 3c). Furthermore, LET7TPR associated with LET7PPase in co-immunoprecipitation (Co-IP) assays (Fig. 3d) and interacted with LET7PPase in yeast two-hybrid (Y2H) assays (Fig. 3e) and in vitro pull-down assays (Fig. 3f). To determine the binding kinetics and affinity between LET7PPase and LET7TPR, we performed biolayer interferometry (BLI) assays by immobilizing glutathione S-transferase (GST)–LET7TPR on the sensor chip surface and injecting a series of concentrations of soluble hexahistidine (HIS)–LET7PPase proteins. BLI sensor grams showed that LET7PPase bound to LET7TPR with a dissociation constant (Kd) of 6.086 µM, but not to another TPR domain protein, TPR5G (Fig. 3g and Extended Data Fig. 3a).
The crystal structure and AlphaFold-based analysis of LET7 revealed the interface between LET7PPase and LET7TPR mediated by several hydrogen bonds25 (Extended Data Fig. 3b). A hydrogen bond between Lys59 in LET7TPR and Gln440 in LET7PPase stabilizes the intramolecular interaction of LET7 (Fig. 3h)25. To explore the functional significance of this interaction, we altered Lys59 of LET7 to aspartic acid (LET7(K59D)) and Gln440 to lysine (LET7(Q440K)), which individually disrupt the Lys59–Gln440 hydrogen bond. The LET7(K59D) or LET7(Q440K) single mutant relieved LET7 autoinhibition, resulting in increased phosphatase activities. However, the double mutant LET7(K59D/Q440K), which could potentially reform the hydrogen bond between K59D and Q440K, displayed phosphatase activities similar to LET7, indicating the restored interaction between LET7TPR and LET7PPase, thereby inhibiting LET7 activities (Fig. 3i). Taken together, these data support the idea that LET7TPR and LET7PPase engage in an intramolecular interaction suppressing LET7PPase phosphatase activity, which otherwise activates SUMM2-dependent autoimmunity.
HOP1 interacts with LET7 and is involved in mekk1 autoimmunity
To uncover the regulatory mechanisms of LET7, we employed a Y2H screen using LET7 as the bait against an Arabidopsis cDNA library to identify LET7-interacting proteins. Among in-frame interacting proteins, several candidates encode protein chaperones or co-chaperones that regulate protein folding and stability (Extended Data Fig. 3c). Among them, alterations in HOP1, including three alleles of T-DNA insertional mutants, hop1-1, hop1-2 and hop1-3, substantially suppressed the RNAi-MEKK1-triggered growth defects (Fig. 4a,b and Extended Data Fig. 3d). Additionally, cell death, H2O2 accumulation (Fig. 4c) and elevated PR1 expression (Fig. 4d) triggered by RNAi-MEKK1 were compromised in hop1-2 compared with WT plants. Growth defects, cell death (Fig. 4e) and the increased expression of PR1 and PR2 (Fig. 4f) in mekk1 were markedly alleviated in mekk1 hop1-2 (Extended Data Fig. 3e). Moreover, transgenic plants expressing HOP1 tagged with GFP under the 35S promoter (p35S::HOP1-GFP) restored growth defects (Fig. 4g) and PR1 gene accumulation (Fig. 4h) triggered by RNAi-MEKK1 in hop1-2. The mutant of required for Mla12 resistance 1 (rar1-21), which encodes a co-chaperone complexing with HSP90 required for the function of multiple NLRs34,35,36, did not obviously affect growth defects triggered by RNAi-MEKK1 at 22–23 °C (Extended Data Fig. 3f), which is consistent with a previous report11.
Fig. 4: HOP1 interacts with LET7 in regulating mekk1 cell death.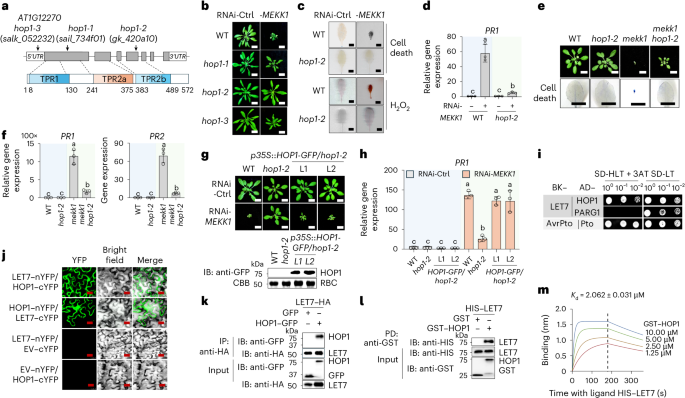
a, Schematic diagram of mutant lines and protein motifs of HOP1. Top: T-DNA insertions in HOP1 (AT1G12270) for three mutant lines are shown with three T-DNA insertional lines. The open boxes are 5′ and 3′ UTRs, the grey boxes indicate exon protein-coding regions and the lines indicate introns. Bottom: protein motifs, including TPR1, TPR2a and TPR2b, with amino acid positions, are labelled. b–d, The hop1 mutants suppress RNAi-MEKK1-triggered growth defects, cell death, H2O2 production and PR1 gene expression. WT and hop1 plants are shown three weeks after inoculation with Agrobacterium carrying the VIGS vector targeting GFP as a control (Ctrl) or RNAi-MEKK1 (b). Scale bars, 1 cm. Detached leaves were stained with trypan blue for cell death or DAB for H2O2 accumulation (c). Scale bars, 0.5 cm. The expression of PR1 was normalized to that of ACTIN2, and the data are shown as means ± s.d. (n = 3, biologically independent samples) (d). Column 1 versus Column 2, P < 0.0001, Column 1 versus Column 3, P > 0.9999, Column 3 versus Column 4, P = 0.0222. e,f, The hop1-2 mutant suppresses growth defects, cell death and the elevated expression of PR1 and PR2 in mekk1. Three-week-old soil-grown plants are shown (e, top) with leaves stained by trypan blue for cell death (e, bottom). Scale bars, 1 cm. The expression of PR1 and PR2 was normalized to ACTIN2. PR1: Column 1 versus Column 3, P < 0.0001, Column 3 versus Column 4, P < 0.0001, Column 2 versus Column 4, P = 0.0003; PR2: Column 1 versus Column 3, P < 0.0001, Column 3 versus Column 4, P < 0.0001, Column 2 versus Column 4, P = 0.0012. g,h, Expression of HOP1–GFP restores RNAi-MEKK1-induced growth defects and PR1 expression in hop1-2. HOP1 tagged with GFP under the 35S promoter (p35S::HOP1-GFP) was transformed into hop1-2. Scale bars, 1 cm (g, top). Immunoblotting by an anti-GFP antibody shows HOP1–GFP proteins with CBB staining RBC as a loading control (g, bottom). The expression of PR1 was normalized to ACTIN2 (h). Column 1 versus Column 2, P > 0.9999, Column 1 versus Column 3, P > 0.9999, Column 1 versus Column 5, P < 0.0001, Column 2 versus Column 3, P > 0.9999, Column 2 versus Column 4, P > 0.9999, Column 5 versus Column 6, P < 0.0001, Column 6 versus Column 7, P < 0.0001, Column 6 versus Column 8, P < 0.0001. i, LET7 interacts with HOP1 in Y2H assays. The experiments were performed similarly to those in Fig. 3e. j, LET7 associates with HOP1 in BiFC assays. LET7 or HOP1 was fused with the N-terminal or C-terminal half of YFP (LET7–nYFP, LET7–cYFP, HOP1–cYFP or HOP1–nYFP) and co-expressed in N. benthamiana leaves with empty vectors carrying nYFP or cYFP (EV–nYFP or EV–cYFP) as controls. Signals were observed via confocal microscopy at 48 h post-inoculation. Scale bars, 25 μm. k, LET7 associates with HOP1 in Co-IP assays. LET7–HA was co-expressed with HOP1–GFP or GFP in protoplasts. Total proteins were immunoprecipitated with anti-HA affinity beads, followed by immunoblotting with an anti-GFP or anti-HA antibody (top two panels). Proteins before immunoprecipitation were immunoblotted and are shown as input controls (bottom two panels). l, LET7 interacts with HOP1 in pull-down assays. GST or GST–HOP1 immobilized on glutathione agarose was incubated with HIS–LET7 proteins. Top: washed beads were pelleted for immunoblotting using an anti-HIS antibody. Middle and bottom: input proteins are shown with immunoblotting before pull-down. m, LET7 interacts with HOP1 in BLI assays. Recombinant HIS–LET7 proteins were immobilized on Ni-NTA biosensor chips and incubated over a range of concentrations (1.25–10 μM) of soluble GST–HOP1 proteins as the analyte. The plot shows the association (0 to 180 s) and dissociation (180 to 360 s) times of the interaction. The equilibrium Kd of the LET7 and HOP1 interaction is 2.062 ± 0.031 μM as determined by Octet BLI analysis software. The data are shown as mean ± s.d. (n = 3 biologically independent samples in d, f and h). Different letters indicate significant differences determined by one-way analysis of variance followed by Tukey’s test (P < 0.05). The experiments were repeated four times in b, e, i, l and m and three times in c, d, f–h, j and k with similar results.
HOP1 has three TPR domains, TPR1, TPR2a and TPR2b, and belongs to the conserved TPR family of co-chaperones of HSP70 and HSP90 (refs. 26,27) (Fig. 4a). Y2H assays confirmed the interaction between LET7 and HOP1 (Fig. 4i). Additionally, bimolecular fluorescence complementation (BiFC) assays co-expressing LET7 and HOP1 fused with either the C-terminal or N-terminal half of yellow fluorescent protein (YFP), but not the vector controls, reconstituted strong YFP signals in N. benthamiana (Fig. 4j). Moreover, Co-IP and pull-down assays indicated that LET7 interacted with HOP1 in vivo and in vitro (Fig. 4k,l). BLI assays further revealed the high affinity between LET7 and HOP1 with a Kd of 2.062 μM (Fig. 4m). These data collectively demonstrate that HOP1 interacts with LET7 and is required for mekk1-triggered cell death.
HOP1 interacts with LET7TPR and releases the autoinhibition of LET7 phosphatase activity
We further mapped the interaction domains of LET7 and HOP1 via Y2H assays using a series of truncation variants (Extended Data Fig. 3g). LET7 interacted strongly with HOP1TPR1 and HOP1TPR1/2a, but weakly with HOP1TPR2a/2b and HOP1TPR2b. LET7TPR, but not LET7PPase, interacted strongly with HOP1TPR1 and HOP1TPR1/2a. Conversely, LET7PPase, but not LET7TPR, weakly interacted with HOP1TPR2a/2b and HOP1TPR2b (Extended Data Fig. 3g). The LET7 and HOP1 interaction is thus predominantly mediated by the LET7TPR and HOP1TPR1 domains. In line with this observation, HOP1TPR1–HA co-immunoprecipitated with LET7TPR–FLAG (Fig. 5a).
Fig. 5: HOP1 interacts with LET7TPR and relieves LET7TPR-mediated suppression of LET7PPase phosphatase activity and cell death.
a, HOP1TPR1 associates with LET7TPR in Co-IP assays in protoplasts. The experiments were performed similarly to those in Fig. 3d. b, HOP1 alleviates the inhibitory effect of LET7TPR on the phosphatase activity of LET7PPase. The experiments were performed similarly to those in Fig. 2e. The same amount of GST proteins was added as a control for HOP1. c, HOP1 reduces LET7TPR and LET7PPase interaction in pull-down assays. Glutathione sepharose beads containing GST–LET7PPase and HIS–LET7TPR were incubated with or without HIS–SUMO–HOP1. Top: washed beads were pelleted for immunoblotting using an anti-HIS antibody. Bottom: input proteins are shown by CBB staining. d, LET7TPR binds to HOP1 in BLI assays. Recombinant GST–LET7TPR proteins were immobilized on GST biosensor chips and incubated over a range of concentrations (2.5–20 μM) of soluble HIS–HOP1 proteins as the analyte. e, The conserved regulation of PP5 by HOP1 across plants and humans. PP5 phosphatase activities in different combinations of Arabidopsis LET7 and HOP1 as well as human HsPP5 and HsHOP are shown. The experiments were performed similarly to those in Fig. 2e. f, AlphaFold2-multimer structural prediction of the LET7TPR–HOP1TPR1 complex. A close-up view of the protein interface shows notable interacting residues in the stick representation. The interface reveals that LET7TPR–LET7PPase-interacting residues such as Tyr63, Lys59 and His27 are involved in putative electrostatic interactions with HOP1. The dashed lines indicate electrostatic interactions. g, The binding of LET7(K59D) with HOP1 in BLI assays. Recombinant GST–HOP1 proteins were immobilized on GST biosensor chips and incubated over a range of concentrations (1.25–10 μM) of soluble HIS–LET7(K59D) proteins as the analyte. h, HOP1(D100K/E104K) reduces interaction with LET7 in Co-IP assays in protoplasts. The indicated constructs were co-expressed in Arabidopsis protoplasts. Total proteins were immunoprecipitated with GFP-Trap beads and immunoblotted with an anti-HA or anti-GFP antibody (top two panels). Protein inputs are shown with immunoblotting before immunoprecipitation (bottom two panels). i, HOP1(D100K/E104K) reduces interaction with LET7 in in vitro pull-down assays. Equal amounts of HIS–HOP1, HIS–HOP1(D100K) and HIS–HOP1(D100K/E104K) proteins were co-incubated with GST–LET7 glutathione beads for the pull-down assay. Top: washed beads were pelleted for immunoblotting using an anti-HIS antibody. Bottom: input proteins are shown by CBB staining. j, HOP1(D100K/E104K) interaction with LET7 in BLI assays. Recombinant GST–HOP1(D100K/E104K) proteins were immobilized on GST biosensor chips and incubated over a range of concentrations (1.25–10 μM) of soluble HIS–LET7 proteins as the analyte. k, HOP1(D100K/E104K) reduces HOP1 activation of LET7 phosphatase activities. Equal amounts of HIS-tagged HOP1, HOP1(D100K), HOP1(E104K) or HOP1(D100K/E104K) proteins were co-incubated with GST–LET7 for the phosphatase assay. l, Dynamics of HOP1–LET7 association upon RNAi-MEKK1 and expression of MPK4. Protoplasts isolated from plants two weeks after VIGS-MEKK1 or Ctrl were transfected with HOP1–GFP and LET7–FLAG, along with or without MPK4–HA or MPK4ac–HA. Total proteins were immunoprecipitated with GFP-Trap beads followed by immunoblotting with an anti-FLAG or anti-GFP antibody (top two panels). Proteins before immunoprecipitation were immunoblotted and are shown as input controls (bottom three panels). The experiments were repeated three times with similar results. The data are shown as mean ± s.d. (n = 3 biologically independent samples in b, e and k).
Next, we tested whether HOP1–LET7TPR interaction could affect LET7 phosphatase activity. Consistent with the inhibitory function of LET7TPR, LET7 exhibited a considerably weaker phosphatase activity than LET7PPase (Fig. 5b and Extended Data Fig. 4a,b). Co-incubation with HOP1 substantially increased LET7 phosphatase activity (Fig. 5b). Furthermore, HOP1 markedly de-repressed LET7TPR-mediated inhibition of LET7PPase phosphatase activity (Fig. 5b). HOP1 did not affect LET7PPase phosphatase activity (Extended Data Fig. 4c). These results suggest that HOP1 interacts with LET7TPR, alleviating the LET7TPR suppression of LET7PPase phosphatase activity. We further examined whether HOP1 binding to LET7TPR may block the LET7TPR–LET7PPase intramolecular interaction. Indeed, HOP1 reduced the pull-down efficiency between LET7PPase and LET7TPR (Fig. 5c), and HOP1TPR1 also reduced the LET7PPase–LET7TPR association in Co-IP assays (Extended Data Fig. 4d). Furthermore, the HOP1 and LET7TPR binding affinity (Kd = 1.862 µM) (Fig. 5d) was stronger than the LET7PPase and LET7TPR intramolecular interaction (Kd = 6.086 µM) (Fig. 3g), supporting the idea that HOP1 binds to LET7TPR in suppressing the LET7 intramolecular interaction.
The human HOP1 homologue, HsHOP/STIP1 (Stress-induced phosphoprotein 1), also enhanced the phosphatase activity of HsPP5 (Fig. 5e). In addition, Arabidopsis HOP1 de-repressed the HsPP5 activity, and conversely, HsHOP released Arabidopsis LET7 autoinhibition (Fig. 5e). Moreover, HsHOP bears a strong binding affinity to HsPP5 in BLI assays (Extended Data Fig. 4e). Our data thus indicate a conserved regulatory mechanism and interchangeable roles of HOP and PP5 in plants and humans, in line with highly conserved PP5 phosphatases and HOP1 co-chaperones in eukaryotes26,27,28,29.
The polar interaction interface between HOP1 and LET7TPR enables relieving LET7 autoinhibition
The AlphaFold-predicted structure of HOP1 resembles the crystal structures of yeast and human co-chaperone HOP1s (Extended Data Fig. 4f)26,37. The predicted LET7TPR–HOP1TPR1 complex revealed several putative polar interactions, including a salt bridge between LET7TPR Lys59 and HOP1TPR1 Asp100, and hydrogen bonds between LET7TPR Tyr63 and HOP1TPR1 Trp69 and between LET7TPR His27 and the peptide backbone of HOP1 (Fig. 5f). In addition, acidic amino acids LET7TPR Glu61 and HOP1TPR1 Glu104 are involved in electrostatic interactions with the N termini of helices in the interaction interface (Fig. 5f). Importantly, these residues, including His27, Lys59 and Tyr63 in LET7TPR mediating polar interactions with HOP1, reside on the key interaction interface with LET7PPase in the crystal structure of LET7/AtPAPP5 (PDB ID: 7OBE)25, further substantiating the idea that HOP1 displaces LET7TPR from LET7PPase to relieve the autoinhibition of LET7 catalytic activity.
To examine the biological function of the HOP1–LET7 interaction, we generated LET7(K59D), where a positively charged residue was changed to a negative charge. Compared with LET7, LET7(K59D) showed reduced interaction with HOP1 in pull-down assays (Extended Data Fig. 4g). Furthermore, BLI assays quantified the decreased binding affinity of HOP1 to LET7(K59D) (Kd = 13.705 µM) (Fig. 5g) compared with LET7 (Kd = 2.062 µM) (Fig. 4m). Conversely, we also generated HOP1 mutants, including HOP1(D100K), HOP1(E104K) and HOP1(D100K/E104K), to switch negative charges to positive charges in the LET7TPR interaction interface. LET7 displayed a slightly reduced association with HOP1(D100K) and HOP1(E104K) and a much-decreased association with HOP1(D100K/E104K) compared with HOP1 in Co-IP assays (Fig. 5h). Similarly, pull-down assays also showed decreased LET7 interaction with HOP1(D100K) and HOP1(D100K/E104K) compared with HOP1 (Fig. 5i). BLI assays further quantified the Kd value of 24.182 μM for the LET7–HOP1(D100K/E104K) interaction (Fig. 5j). Consistent with the reduced interaction with LET7, HOP1(D100K/E104K) was compromised in de-repressing LET7 phosphatase activity (Fig. 5k). These data thus highlight the essential role of the interaction interface between HOP1 and LET7TPR in releasing the autoinhibition of LET7 phosphatase activity.
We subsequently investigated the HOP1–LET7 interaction by genetically interfering with the MEKK1–MKK1/2–MPK4 cascade. In RNAi-MEKK1 plants, the association between HOP1 and LET7 was increased compared with RNAi-Ctrl plants (Fig. 5l). Conversely, the active form of MPK4 (MPK4ac) suppressed the HOP1–LET7 association in RNAi-MEKK1 plants (Fig. 5l and Extended Data Fig. 5a). The MEKK1–MKK1/2–MPK4 cascade represses MEKK2 expression, which otherwise triggers SUMM2-dependent autoimmunity15,16. Consistently, overexpression of MEKK2 enhanced the HOP1–LET7 interaction (Extended Data Fig. 5a). The Pseudomonas syringae HopAI1 effector hijacks the MEKK1–MKK1/2–MPK4 cascade for virulence9. Expression of HopAI1 in planta increased HOP1–LET7 association (Extended Data Fig. 5b). Together, these findings support the idea that the MEKK1–MKK1/2–MPK4 cascade inhibits the HOP1–LET7 interaction, maintaining a basal LET7 phosphatase activity. Disruption of this cascade, for instance by P. syringae effector HopAI1, alleviates LET7 autoinhibition and promotes the HOP1–LET7 interaction, ultimately elevating LET7 phosphatase activity to trigger autoimmunity.
LET7 interacts with and dephosphorylates CRCK3
We next investigated the potential target(s) of LET7 as a phosphatase in activating SUMM2-mediated autoimmunity. Several kinases have been implicated in this process, including the MEKK1–MKK1/2–MPK4 cascade, malectin-like receptor kinases LET1 and LET2 and cytosolic kinase CRCK3 (refs. 10,19,20,21,22,38). Notably, flg22-triggered MAPK activation in let7-1, let7-2 and let7-3 mutants was similar to that in WT plants (Extended Data Fig. 6a), implying that LET7 may not interfere with MAPK activation. LET2 promotes LET1 phosphorylation as detected with a protein mobility shift21. Expression of LET7 did not obviously affect LET2-induced LET1 phosphorylation, suggesting that LET7 may not regulate the LET2–LET1 phosphorylation cascade (Extended Data Fig. 6b).
When CRCK3–GFP was expressed in N. benthamiana, it exhibited two discrete bands in immunoblots (Fig. 6a). Importantly, co-expression of LET7 reduced the ratio of the upper to lower bands of CRCK3. The upper band was abolished by treatment with K252a, a general kinase inhibitor, or calf intestinal alkaline phosphatase, suggesting phosphorylated CRCK3 proteins (Fig. 6a). Similarly, CRCK3–FLAG displayed two bands in Arabidopsis protoplasts (Fig. 6b). Remarkably, LET7–HA, but not the catalytically inactive LET7(H290N) mutant, reduced the ratio of the upper to lower bands of CRCK3–FLAG when we used proteins immunoprecipitated from protoplasts (Fig. 6b). Moreover, the LET7(W372F) phosphatase hypermutant eliminated the upper band of CRCK3–FLAG (Fig. 6b). These data indicate that LET7 mediates CRCK3 dephosphorylation in planta.
Fig. 6: LET7 dephosphorylates CRCK3 in an HOP1-dependent manner.
a, LET7 reduces CRCK3 phosphorylation. A. tumefaciens cultures carrying p35S::CRCK3-GFP with or without Dex-inducible LET7-HA were infiltrated into N. benthamiana leaves. Two days post-inoculation, infiltrated leaf areas were treated with 1 µM K252a, H2O or 10 μM Dex and harvested after 24 h. Calf intestinal alkaline phosphatase (CIP) was added to lysed cells in the indicated sample. Total proteins were immunoblotted using an anti-GFP antibody to detect CRCK3–GFP expression (top) or anti-HA antibody to detect LET7–HA (middle). Signal intensity ratios of phosphorylated CRCK3 (pCRCK3) to CRCK3 were calculated, with ImageJ (version imagej.js v0.6.0) (ref. 58) used for band intensity quantification. The ratio in the first lane was set to 1. Bottom: CBB-stained RBC served as a loading control. b, Compared with LET7, LET7(W372F) displays increased activity but LET7(H290N) displays reduced activity in de-phosphorylating CRCK3. CRCK3–FLAG expressed in protoplasts was purified with anti-FLAG beads. LET7–HA, LET7(H290N)–HA or LET7(W372F)–HA expressed from protoplasts was isolated via anti-HA agarose and co-incubated with CRCK3–FLAG beads in a phosphatase buffer at 30 °C for 90 min. Top: CRCK3–FLAG was detected by immunoblotting using an anti-FLAG antibody. The quantification was performed similarly to that in a. The bottom two panels show the inputs of CRCK3–FLAG and LET7–HA proteins before co-incubation. c, Purified LET7 proteins dephosphorylate CRCK3. CRCK3–GFP proteins expressed in transgenic plant seedlings were immunoprecipitated using GFP-Trap beads and incubated with HIS-tagged LET7, LET7(H290N) or LET7(W372F) proteins purified from E. coli for the indicated times. CRCK3–GFP proteins eluted from GFP-Trap beads were immunoblotted with an anti-pSer/Thr antibody (IB: anti-pS/T). The quantifications show the relative signal intensity ratios compared with time 0, which was set as 1. CBB staining shows LET7 proteins. d, LET7 interacts with CRCK3 in BiFC assays. LET7 or CRCK3 fused with the N- or C-terminal half of YFP was co-expressed in different combinations or with an empty vector (EV) in N. benthamiana leaves. Images were observed using a confocal microscope at 48 h post-inoculation. Scale bars, 25 μm. e, LET7 associates with CRCK3 in Co-IP assays. LET7–HA was co-expressed with GFP or CRCK3–GFP in protoplasts. Total proteins were immunoprecipitated with GFP-Trap beads and immunoblotted with an anti-HA or anti-GFP antibody (top two panels), with inputs shown with immunoblotting before immunoprecipitation (bottom two panels). f, LET7TPR and LET7PPase domains associate with CRCK3 in pull-down assays. GST–CRCK3 or GST immobilized on glutathione sepharose was incubated with HIS–LET7TPR or HIS–LET7PPase and pelleted for immunoblotting with an anti-HIS antibody (top) with CBB staining for input proteins (bottom). g, HOP1 enhances LET7-mediated CRCK3 dephosphorylation. CRCK3–FLAG was co-expressed with a vector control (−) or LET7–HA together with GFP or HOP1–GFP in N. benthamiana leaves for three days. Total proteins were immunoblotted with an anti-FLAG (top), anti-HA (middle) or anti-GFP (bottom) antibody. The quantification of CRCK3 phosphorylation was calculated as in a. h, HOP1 is required for LET7-mediated dephosphorylation of CRCK3. CRCK3–FLAG was expressed with a vector control (−), LET7–HA or HOP1–GFP in WT and hop1-2 protoplasts. Total proteins were immunoblotted with an anti-FLAG, anti-HA or anti-GFP antibody. The quantification of CRCK3 phosphorylation was calculated as in a. i, RNAi-MEKK1 induces LET7-dependent CRCK3 dephosphorylation. Two independent homozygous lines of p35S::CRCK3-GFP expressed in the WT (L1 and L2) or let7-2 (L3 and L4) were used for VIGS assays. Top: two weeks after inoculation, total proteins were subjected to immunoblotting with an anti-GFP antibody. The quantification of CRCK3 phosphorylation was calculated as in a. Bottom: CBB-stained RBC served as the loading control. j, Overexpressing LET7 triggers growth defects in p35S::CRCK3-GFP plants. p35S::LET7-FLAG was transformed into p35S::CRCK3-GFP or WT plants. Four-week-old soil-grown plants were photographed. Among 103 T1 transgenic plants carrying p35S::LET7-FLAG p35S::CRCK3-GFP, 34 lines (33%) were severe dwarfs with dark leaves (Group 1), 43 (42%) were small (Group 2) and 26 (25%) were comparable to p35S::LET7-FLAG and p35S::CRCK3-GFP transgenic plants (Group 3). Scale bars, 1 cm. k, Dwarf transgenic plants carrying p35S::LET7-FLAG p35S::CRCK3-GFP showed elevated PR1 protein expression. Top: PR1 was detected by immunoblotting using an anti-PR1 antibody. CRCK3–GFP and LET7–FLAG were detected by immunoblotting with an anti-GFP and anti-FLAG antibody, respectively (middle two panels), with CBB-stained RBC as a loading control (bottom). l, Co-expressing LET7 and CRCK3 triggers cell death in N. benthamiana. p35S::CRCK3-HA was co-expressed with an empty vector control (−) or Dex::LET7-HA in N. benthamiana leaves for two days, followed by inoculation with 10 μM Dex. Images were taken under UV light one day after Dex treatment. Immunoblots using an anti-HA antibody show LET7–HA and CRCK3–HA protein expression with CBB-stained RBC as a loading control. The experiments were repeated at least three times with similar results.
To assess whether LET7 directly dephosphorylates CRCK3, we used LET7, LET7(W372F) or LET7(H290N) proteins purified from E. coli and CRCK3–GFP immunoprecipitated from p35S::CRCK3-GFP transgenic plants. Compared with LET7(H290N), LET7 reduced the level of phosphorylated CRCK3–GFP, as detected by an anti-pSer/Thr antibody (Fig. 6c). The dephosphorylation of CRCK3–GFP was further enhanced by LET7(W372F) (Fig. 6c). These data substantiate the idea that LET7 directly dephosphorylates CRCK3. In line with these observations, LET7 associated with CRCK3 in BiFC assays (Fig. 6d) and Co-IP assays (Fig. 6e). Similar to LET7, LET7(H290N) still interacted with CRCK3 (Extended Data Fig. 6c). LET7PPase had a stronger interaction with CRCK3 than LET7TPR did in pull-down and Y2H assays (Fig. 6f and Extended Data Fig. 6d).
Given that HOP1 interacted with and promoted LET7 phosphatase activity (Fig. 5), we next examined whether HOP1 enhanced LET7-mediated dephosphorylation of CRCK3. HOP1 markedly augmented LET7-mediated dephosphorylation of CRCK3 in N. benthamiana (Fig. 6g). Similarly, HOP1 promoted LET7-mediated dephosphorylation of CRCK3 in Arabidopsis protoplasts (Fig. 6h). LET7-mediated dephosphorylation of CRCK3 was attenuated in hop1-2, which could be restored by expressing HOP1 (Fig. 6h), further supporting the role of HOP1 in activating LET7 phosphatase activity.
LET7-mediated CRCK3 dephosphorylation is associated with mekk1 autoimmunity
We further explored the biological relevance of LET7-mediated CRCK3 dephosphorylation. Upon the onset of autoimmune signalling by RNAi-MEKK1, the ratio of phosphorylated to unphosphorylated CRCK3–GFP proteins was substantially reduced compared with RNAi-Ctrl in p35S::CRCK3-GFP WT transgenic plants (Fig. 6i, left). In contrast, the reduced CRCK3 phosphorylation upon MEKK1 silencing was much less pronounced in the let7-2-background transgenic plants (Fig. 6i, right). Additionally, expression of bacterial HopAI1 in planta promoted CRCK3 dephosphorylation (Extended Data Fig. 6e). HopAI1 induces SUMM2-dependent growth defects and cell death9. HopAI1-triggered growth defects were substantially attenuated in let7-2 compared with the WT (Extended Data Fig. 6f), suggesting that LET7-mediated CRCK3 dephosphorylation is associated with SUMM2 activation upon disruption of the MEKK1–MKK1/2–MPK4 cascade by HopAI1. This was further corroborated by the impaired CRCK3 phosphorylation in the mpk4 mutant20.
We next tested whether LET7 could promote CRCK3 in triggering cell death. We introduced p35S::LET7-FLAG into p35S::CRCK3-GFP transgenic plants displaying normal growth phenotypes. Among 103 T1 transgenic plants carrying both p35S::LET7-FLAG and p35S::CRCK3-GFP transgenes, 34 exhibited severe growth defects (33%), 43 showed moderate growth defects (42%) and 26 resembled WT plants (25%) (Fig. 6j). Additionally, elevated PR1 levels were correlated with severe growth defects of transgenic plants (Fig. 6k). Moreover, dexamethasone (Dex)-induced expression of LET7-HA (Dex::LET7-HA) triggered cell death when co-expressed with CRCK3-HA, but not when expressed alone (Fig. 6l). Taken together, these data support the idea that LET7-mediated dephosphorylation of CRCK3 leads to autoimmunity and cell death.
LET7 and HOP1 stabilize SUMM2 in activating cell death
The expression of an active form of SUMM2 (SUMM2ac, D478V in the MHD motif)9 under the 35S promoter (p35S::SUMM2ac-HA) in WT plants resulted in variable degrees of growth defects (three groups, G1–G3) correlated with SUMM2ac protein levels (Fig. 7a). The severity of growth defects of p35S::SUMM2ac-HA transgenic plants was mitigated in let7-2 (Fig. 7a). Representative plants from G1, exhibiting mild growth defects, showed reduced SUMM2ac levels in let7-2 compared with the WT (Fig. 7b). These findings suggest that LET7 may stabilize SUMM2. Indeed, an increased inoculum of Agrobacterium carrying LET7-FLAG with the same amount of Agrobacterium carrying SUMM2-HA led to increased SUMM2 protein levels in N. benthamiana (Fig. 7c). LET7TPR, but not LET7PPase, elevated the protein levels of SUMM2 and SUMM2ac in protoplasts (Fig. 7d and Extended Data Fig. 7a). In contrast, LET7TPR did not affect the protein levels of other NLRs, including RPM1 and RPS2 (Extended Data Fig. 7b). Taken together, these data suggest that LET7 modulates SUMM2 protein levels via its TPR domain.
Fig. 7: LET7 and HOP1 regulate the stability of SUMM2 in controlling cell death.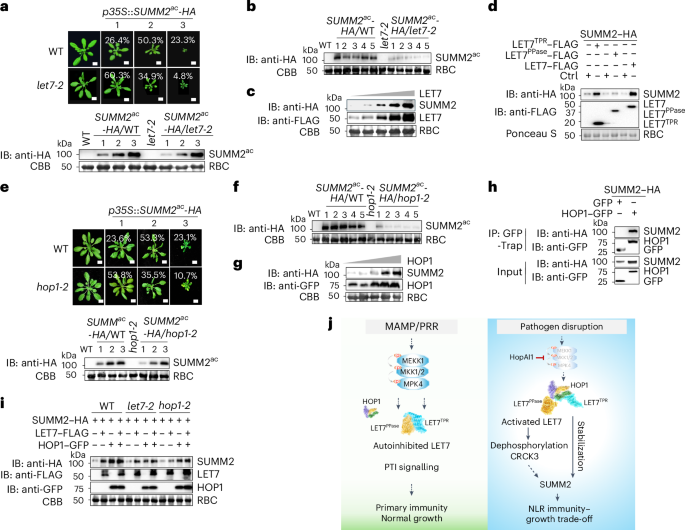
a, SUMM2ac-triggered growth defects are alleviated in let7-2. Four-week-old soil-grown T1 transgenic plants carrying p35S::SUMM2ac-HA in the WT and let7-2 were grouped into three categories (1, 2 and 3) on the basis of the level of dwarfism. The percentage of transgenic plants in each category is shown with 137 plants of p35S::SUMM2ac-HA WT and 126 plants of p35S::SUMM2ac-HA let7-2. Scale bars, 1 cm. SUMM2ac–HA proteins are shown by immunoblotting using an anti-HA antibody with CBB-stained RBC as a loading control. b, SUMM2ac–HA protein levels are reduced in let7-2 compared with the WT. Total proteins of five randomly selected G1 plants of p35S::SUMM2ac-HA in the WT and let7-2 were immunoblotted using an anti-HA antibody with CBB-stained RBC as a loading control. c, LET7 enhances SUMM2 protein accumulation. Equal amounts of A. tumefaciens cultures (OD600 = 0.6) carrying p35S::SUMM2-HA were co-infiltrated with A. tumefaciens cultures carrying p35S::LET7-FLAG at different concentrations (OD600 = 0.1, 0.25, 0.5, 0.75 and 0.9) into N. benthamiana leaves. A. tumefaciens cultures carrying the empty vector were used to adjust the final bacterial concentration to be equal across all combinations. SUMM2–HA proteins were detected using an anti-HA antibody (top), and LET7–FLAG proteins were detected using an anti-FLAG antibody (middle), with CBB-stained RBC as a loading control (bottom). d, Expression of LET7TPR, but not LET7PPase, enhances SUMM2 protein levels. SUMM2–HA was co-expressed with a vector control (Ctrl), LET7TPR–FLAG, LET7PPase–FLAG or LET7–FLAG in protoplasts. Total proteins were immunoblotted with an anti-HA antibody to detect SUMM2–HA (top) and anti-FLAG antibody for LET7–FLAG (middle) with Ponceau-S-stained RBC as a loading control (bottom). e, SUMM2ac-triggered cell death is compromised in hop1-2. Four-week-old soil-grown T1 transgenic plants carrying p35S::SUMM2ac-HA in the WT (102 lines) and hop1-2 (97 lines) were grouped into three categories (1, 2 and 3) on the basis of their dwarfism. Scale bars, 1 cm. SUMM2ac–HA proteins are shown by immunoblotting using an anti-HA antibody with CBB-stained RBC as a loading control. f, SUMM2ac–HA protein levels are reduced in hop1-2 compared with the WT. Total proteins of five randomly selected G1 plants of p35S::SUMM2ac-HA in the WT and hop1-2 were immunoblotted using an anti-HA antibody with CBB-stained RBC as a loading control. g, HOP1 enhances SUMM2 protein accumulation. The experiments were performed similarly to those in c using A. tumefaciens cultures carrying HOP1–GFP at varying concentrations. h, HOP1 associates with SUMM2 in Co-IP assays. SUMM2–HA was co-expressed with GFP or HOP1–GFP in protoplasts. Total proteins were immunoprecipitated with GFP-Trap beads followed by immunoblotting using an anti-HA or anti-GFP antibody (top two panels). Proteins before immunoprecipitation are shown as input controls (bottom two panels). i, LET7 and HOP1 independently enhance SUMM2 protein levels. SUMM2–HA was co-expressed with LET7–FLAG, HOP1–GFP or both in WT, let7-2 or hop1-2 protoplasts. Total proteins were immunoblotted with an anti-HA antibody for SUMM2–HA (first panel), anti-FLAG antibody for LET7–FLAG (second panel) and anti-GFP antibody for HOP1–GFP (third panel), with CBB-stained RBC as a loading control (bottom panel). j, A model of the HOP1–LET7 module in maintaining immune homeostasis. Under normal growth conditions, LET7 phosphatase activity remains repressed by the inhibitory interaction between its PPase and TPR domains, supporting plant growth and survival. Plant pattern recognition receptors (PRRs) detect MAMPs, initiating PTI, supporting plant primary immunity. Upon pathogen infection, disruption of the MEKK1–MKK1/2–MPK4 cascade by P. syringae effector HopAI1 leads to enhanced HOP1–LET7 interaction and relieves LET7PPase activity from autoinhibition by LET7TPR. LET7PPase dephosphorylates CRCK3, consequently activating NLR SUMM2-mediated immunity. Additionally, HOP1 and LET7TPR stabilize SUMM2, further sustaining NLR immunity with a plant growth trade-off. The experiments were repeated three times with similar results.
Furthermore, growth defects of p35S::SUMM2ac-HA transgenic plants in hop1-2 were reduced compared with those in the WT (Fig. 7e). SUMM2ac levels in individual G1 transgenic plants were also lower in hop1-2 than in the WT (Fig. 7f). Elevated expression of HOP1, but not other TPR proteins, including TPR14TPR and TPR5G, increased SUMM2 levels in N. benthamiana (Fig. 7g and Extended Data Fig. 7c). In line with this, both HOP1 and LET7 co-immunoprecipitated with SUMM2 (Fig. 7h and Extended Data Fig. 7d), implying that HOP1 and LET7 associate with and stabilize SUMM2. The LET7-mediated SUMM2 stabilization still occurred in hop1-2, and vice versa, HOP1 stabilized SUMM2 in let7-2 (Fig. 7i), implying that LET7 and HOP1 may independently contribute to stabilizing SUMM2 proteins.





