

Abstract
Herpes simplex keratitis (HSK), caused by herpes simplex virus type I (HSV-1) ocular infection, is a leading cause of visual morbidity worldwide, and although cases of HSK can be managed with current medications, new developments are required to make treatments more effective and satisfactory. Current evidence suggests that corneal scarring and vascularization result from chronic inflammation triggered by HSV-1 antigens. The pathogenesis of HSK remains complex and incompletely understood, but there have been many recent advancements have improved our knowledge of HSV-1 and its interactions with the host immune system, particularly in regard to various signaling pathways and regulators. In this review, we discuss the roles of innate immunity in corneal epithelial cells and innate immune cells, DNA sensors and regulators of DNA sensing pathways in HSK caused by acute and recurrent HSV-1 ocular infection and present potential immune-based therapeutic targets for novel HSK treatments.
1 Introduction
Herpes simplex keratitis (HSK) is a leading cause of infectious blindness worldwide (1, 2). HSK is primarily caused through ocular infection by herpes simplex virus type 1 (HSV-1), a member of the Herpesviridae family commonly associated with oral infections (3). Based on data from 2020, the global incidence of HSK is estimated to be 24 cases per 100,000 people, translating to around 1.7 million cases per year (4). HSV-1 is a double-stranded DNA (dsDNA) virus that exhibits a strong neurotropic nature, meaning that it infects and persists in neuronal tissues (3, 5). After primary infection through mucosal or skin contact, HSV-1 travels retrograde along sensory nerve axons to establish latent infection within the trigeminal ganglia (6). During the latent phase, HSV-1 produces latency-associated transcripts (LATs), which maintain the virus’s non-replicative state and prevent host immune clearance (7). HSV-1 can then periodically reactivate, resuming viral replication in response to various stimuli such as stress, fever, ultraviolet (UV) exposure, or immunosuppression (3). The reactivated virus travels anterogradely to the cornea, causing recurrent HSK, which can be split into several clinical subtypes, such as stromal, epithelial, and endothelial, based on the affected corneal layer (1, 8). A study on tree shrews has shown that the HSV-1 viral genes are still active in the corneal and ciliary ganglion tissues even after the acute infection, which demonstrates the complex pathogenesis of this virus since it can have multiple reservoirs (9). Other experimental models are widely used to study HSV-1 keratitis as well, including murine models (10–13), which have been fundamental in dissecting innate and adaptive mechanisms (14); rabbit models (15), which has been used to study corneal latency; and guinea pig models (16), which have provided insights into ocular viral shedding. Because of the high rate of recurrence, several complications may occur, including ulcerations, scarring, and blindness (17). Blindness mainly results from an exaggerated inflammatory response by innate immune cells to HSV-1 infection (18). Innate immunity serves as the first line of defense against HSV-1 (19), playing a critical role in controlling HSK. Understanding the mechanisms by which antiviral innate immunity regulates HSK and how HSV-1 evades these defenses in innate immune cells is essential. Pattern recognition receptors (PRRs), such as DNA sensor cyclic GMP-AMP synthase (cGAS) (20), on innate immune cells detect pathogen-associated molecular patterns (PAMPs) from HSV-1, such as dsDNA, triggering downstream DNA-sensing signaling pathways (19, 21). These pathways recruit innate immune cells, including dendritic cells (DCs), macrophages, natural killer (NK) cells, and neutrophils, to the infection site. These cells secrete inflammatory molecules, promoting effects such as enhanced cell metabolism and further immune cell recruitment (1, 22). Diagnosis of HSK is primarily clinical, and it is usually supplemented with a slit-lamp examination, which uses using a low-power microscope to provide a detailed view of the eye’s structures (8, 23). HSK is typically treated with the antiviral drug acyclovir, which is often supplemented with topical corticosteroids depending on the HSK subtype (24). Alternative approaches for treating HSV-1 include gene-editing strategies, such as mRNA-carrying lentiviral particles delivering SpCas9 mRNA and viral-gene-targeting guide RNAs. These methods have demonstrated inhibition of HSV-1 replication in preclinical studies (2). However, managing HSK remains challenging due to high recurrence rates, immune-mediated corneal damage, and impaired corneal nerve regeneration. Current treatments cannot prevent viral latency or reactivation (8, 25).
2 Classification and pathophysiology of HSK
According to the clinical signs of HSV-1 infection in the cornea, HSK is classified into different clinical types, including epithelial HSK, stromal HSK, and endothelial HSK. Their distinct pathological processes and immune mechanisms are discussed below.
2.1 Epithelial HSK
Epithelial HSK is the most common form of ocular HSV-1 infection and is characterized by active viral replication in the corneal epithelium, which is the outermost layer of the eye’s cornea that plays a vital role in vision and protection, resulting in the destruction of corneal epithelial cells (CECs) (18, 26). It presents as dendritic ulcers, which are superficial corneal ulcers that extend in tree-like patterns, and geographic ulcers, which are a progression of dendritic ulcers and appear as amoeboid-shaped ulcers with scalloped borders (27). The primary symptoms are eye pain, photophobia, tearing, decreased vision, and reduced corneal sensitivity (23). During reactivation, HSV-1 travels from the trigeminal ganglion via the ophthalmic nerve branch and infects corneal epithelial cells, resulting in localized inflammation and corneal scarring (28). PRRs detect dsDNA and other PAMPs from HSV-1, initiating a type I interferon (IFN) response and releasing inflammatory cytokines and chemokines (22, 29–31). The first responders in epithelial HSK are neutrophils, which clear the virus while also causing tissue damage through reactive oxygen species (ROS) (32).
2.2 Stromal HSK
Unlike epithelial HSK, which occurs due to active viral replication, stromal HSK is primarily immune-mediated, meaning that it can occur without detectable viral presence due to the immune system continuing to react even after the virus has been cleared. It is characterized by recurrent inflammation in the corneal stroma, which is the thickest layer of the cornea that provides structural support and facilitates wound healing, and its primary symptoms are scarring, thinning, and vision loss (18, 33). Stromal HSK is a CD4+ T-cell-mediated delayed-type hypersensitivity (DTH) reaction, meaning that even after the initial HSV-1 infection is resolved, CD4+ T cells become activated and secrete pro-inflammatory cytokines, recruiting and activating local macrophages that cause inflammation and tissue damage in the corneal stroma (1, 34).
2.3 Endothelial HSK
Endothelial HSK is characterized by inflammation of the corneal endothelium, the innermost layer of the cornea responsible for nutrient transport and maintaining corneal deturgescence, and it can lead to stromal edema, keratic precipitates, iritis, and elevated intraocular pressure (18, 35). Like stromal HSK, endothelial HSK is primarily immune-mediated and occurs due to a reactive hypersensitivity response to viral antigens in the corneal endothelium that persist even in the absence of live virus (36). Antigen-presenting cells (APCs), such as DCs and macrophages, can migrate to the cornea and express major histocompatibility complex class II (MHC-II) molecules, which activate CD4+ T cells (37). CD4+ T cells infiltrate the posterior stroma and endothelium and produce cytokines in response to residual HSV-1 antigens in the endothelium, activating resident immune cells and leading to the inflammation of the endothelium (38).
3 Role of CECs in HSK
CECs are the outermost layer of cells that cover the front surface of the cornea, and studies have shown that CECs secrete extracellular vesicles whenever the cornea is wounded (39). The immune response of the cornea is predominantly controlled by Anterior Chamber Immune Deviation (ACAID), which prevents the immune system from responding too extremely to various particles, microorganisms, or viruses that enter the eye, which protects the eye from inflammation that can lead to blindness (40). ACAID is initiated when APCs capture antigens in the anterior chamber and migrate to the spleen, where they induce the expansion of regulatory T cells (Tregs). These Tregs secrete immunosuppressive cytokines, such as interleukin-10 (IL-10), which suppress Th1-driven and Th17-driven responses that would otherwise promote neutrophil and macrophages infiltration and corneal scarring (41). By promoting the activity of CD4+ and CD8+ Tregs, ACAID reduces the risk of destructive stromal inflammation by suppressing antigen-specific DTH and effector T cell activity (42). The CECs play an important role in the immune response because they recognize PAMPs and damage-associated molecular patterns (DAMPs), activating neutrophils and causing inflammation (1). After HSV-1 infection, ROS are produced in CECs, which is essential for activating key immune signaling pathways (43). Increased ROS induces Jagged1 (JAG1) expression, and the JAG1-NOTCH1-pULK1 pathway inhibits autophagy and leads to apoptosis of CECs since increased JAG1 leads to the activation of pULK1, which suppresses autophagy and leads to apoptosis (44) (Figure 1). CECs also exhibit antiviral functions, notably through the production of type III IFN. CECs primarily produce type III IFN, which suppresses viral replication and modulates the inflammatory response, and they also produce type I IFN, which activates antiviral mechanisms and recruit immune cells to the site of the infection (45). CECs secrete extracellular vesicles carrying proteins, lipids, and signaling molecules upon injury, which activate neutrophils and initiate inflammation (46). They also produce cytokines, such as IL-18 and IFN-γ, to recruit DCs and macrophages, which process antigens and present them to T cells, activating the adaptive immune response (47).
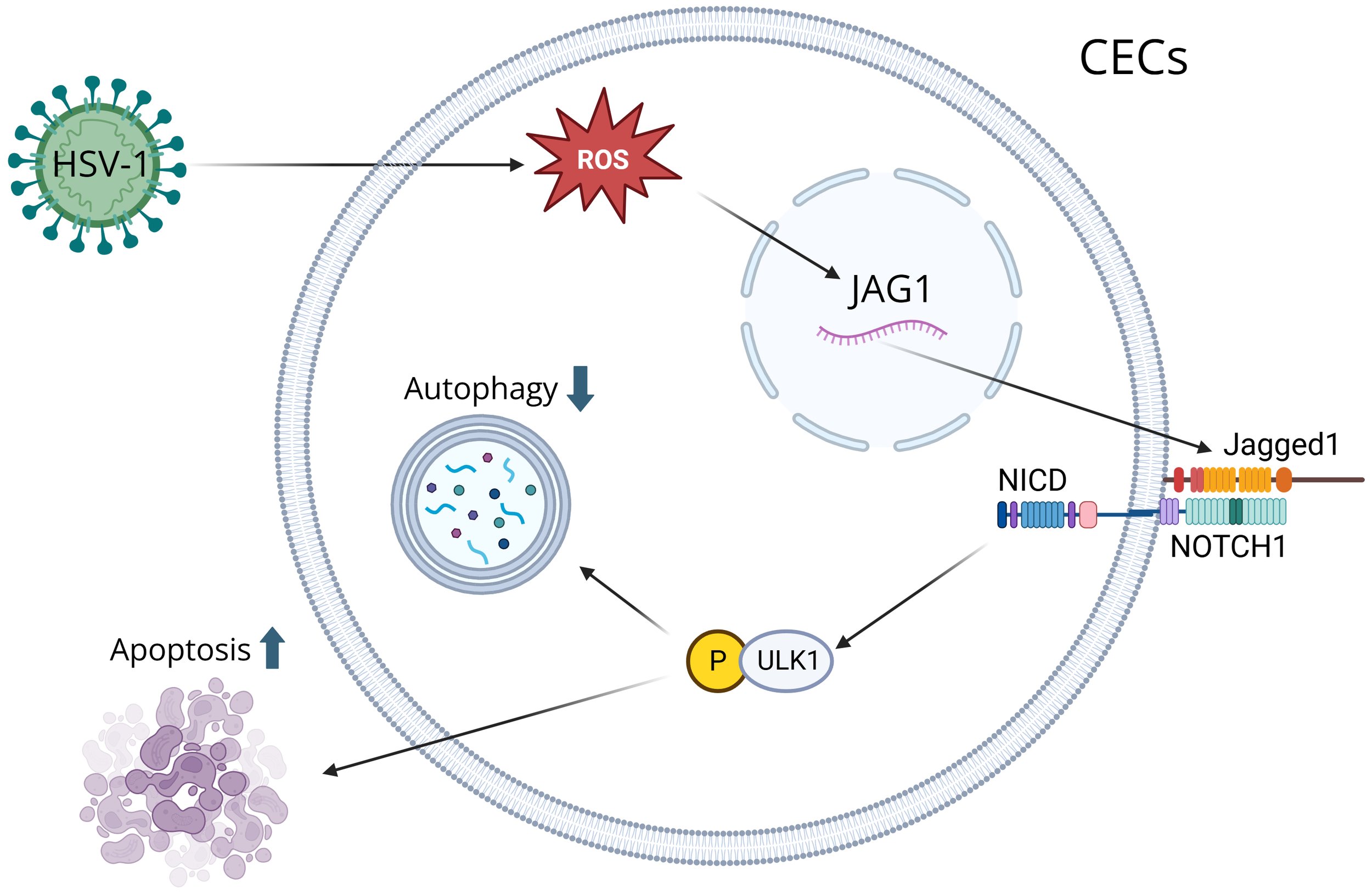
A visual diagram that illustrates how HSV-1 induces corneal epithelial cells (CECs) apoptosis by suppressing autophagy through the ROS-JAG1-NOTCH1-pULK1 signaling pathway. Reactive oxygen species (ROS) are produced during HSV-1 infection, inducing the Jagged1 (JAG1) signaling pathway. ROS modulates JAG1 expression, and when JAG1 binds to a notch receptor on a neighboring cell, it triggers a series of proteolytic cleavages to release the Notch Intracellular Domain (NICD). NICD interacts with ULK1 in the cytoplasm, resulting in its phosphorylation. Through mitochondrial interaction, ULK1 inhibits autophagy and leads to apoptosis.
4 Role of innate immune cells in HSK4.1 DCs
DCs are a special type of antigen-presenting cell that act as the “sentinels” of the immune system and bridge the innate and adaptive immune system by presenting antigens to T cells (48–50). It used to be believed that there were no DCs directly on the cornea, but now it is known that there is a stratified network of DCs throughout the cornea (51). The interesting thing about DCs is that a study has shown that reducing DCs reduces the severity of stromal disease since DCs induce an inflammatory response through the activation of T cells, causing more damage (52). Tripartite Motif 29 (TRIM29) is strongly induced by cytosolic dsDNA in DCs, and TRIM29 deficiency has been shown to increase resistance to HSV-1 through increasing the production of type I IFN (23), suggesting the possible role of TRIM29 in controlling HSK. There are several subsets of DCs present in the peripheral cornea including CD11c+ conventional DCs (cDCs) and plasmacytoid DCs (pDCs). In murine HSV-1 infection, resident cDCs promote local recruitment of NK cells and inflammatory monocytes, which leads to early viral clearance (53). While cDCs promote systemic viral dissemination, resident pDCs play a protective role by limiting viral burden and preserving the function of Tregs, making them extremely important in preventing clinical disease and nerve loss (37). Before infection, many corneal DCs are in an immature state, which supports ACAID. This regulatory environment favors tolerogenic DCs that induce Tregs and dampen inflammation (54). However, upon HSV-1 infection corneal DCs undergo rapid maturation, upregulating MHC-II and producing pro-inflammatory cytokines, thereby promoting the differentiation of effector CD4+ T cells, which contribute to stromal immunopathology (52).
4.2 Macrophages
Macrophages are another type of white blood cell that remove dead cells, kill microorganisms, and stimulate other immune cells (55, 56). Unlike DCs, macrophages are not present in naive corneas, but CCR2+ migratory macrophages are the predominant innate infiltrate within 48 hours, contributing to early viral sensing and cytokine production (57). M1 macrophages are classically activated and produce pro-inflammatory mediators such as IL-6 and TNF-α. These responses promote viral clearance while also causing corneal damage through the recruitment of neutrophils and the amplification of stromal inflammation (58). In contrast, M2 macrophages are alternatively activated and secrete anti-inflammatory mediators like IL-10, which supports tissue repair, resolution of inflammation, and angiogenesis (59). A study has been done that tested a ganglioside GM1 liposome vaccine that encapsulated HSV-1 glycoprotein D and targeted CD169+ macrophages, and the study showed that the vaccine increased the number of corneal infiltrating macrophages, polarizing them toward M1, and there were also significantly more T cells and DCs (10). The Mal adaptor protein plays an important role in TLR9 signaling through ERK1/2 kinases, making it essential for TLR9-mediated expression of IFN-β and TNF-α in macrophages exposed to HSV-1 (60). Macrophages play a key role in the early immune response to HSV-1 in the olfactory epithelium, causing inflammation as the virus spreads from the apical layers to the basal layers and into the underlying tissues (61). Furthermore, the deletion of TRIM18 increases the production of type I IFN response in macrophages, protecting mice from HSV-1 infection (62), suggesting the possible role of TRIM18 in HSK. In addition, overexpression of NOD-like receptor family pyrin domain containing 12 (NLRP12) triggers IL-18-meidated pyroptosis in infected macrophages, amplifying antiviral signaling cascades to alleviate HSK (63).
4.3 Innate lymphoid cells
ILCs are innate lymphocytes that produce cytokines in response to viral infection and inflammation (64). Group 1 ILCs are comprised of noncytotoxic ILC1s and cytotoxic NK cells (65). ILC1s produce IFN-γ in response to IL-12, IL-15, and IL-18, acting as a first line of defense against viral infections (66). Given IFN-γ role as a signature pro-inflammatory cytokine, ILC1s likely stimulate inflammation in response to HSV-1 infection (67). NK cells are a type of white blood cell that can kill their targets autonomously, recognizing and eliminating cells infected with viruses or tumors (68–70). Their recruitment is mediated by chemokines such as CXCL9, CXCL10, and CCL5, which are secreted by infected corneal cells and resident DCs (53). NK cells expressing CD16 can kill HSV-infected cells opsonized with HSV-specific IgG (71). A study has been done that shows that invariant natural killer T (iNKT) cells help protect against HSV-1 because asymptomatic mice had high levels of iNKT1 cells while symptomatic mice had no iNKT cells (72). On the other hand, other studies have shown that NK cells greatly contribute to corneal damage because researchers chemically depleted NK cells in some mice, leading to the severity and frequency of HSK dropping significantly (73). Interestingly, NK cell activity is reduced even though the number of NK cells stays the same in HSK patients, meaning that the impaired function of NK cells might allow HSV-1 to reactivate more easily (74).
4.4 Neutrophils
Neutrophils are another type of white blood cell and act as the first line of defense by engulfing and digesting microorganisms while also releasing enzymes and toxins to kill pathogens and promote inflammation (75). Neutrophils are recruited the earliest either by chemokines such as CXCL1, CXCL2, and CCL3 or by TLR2-myeloid differentiation primary response 88 (MyD88) signaling, which is when HSV-1 glycoproteins via TLR2 induce neutrophil-recruiting chemokines (76). Through phagocytosis, degranulation, and the release of antiviral cytokines and neutrophil extracellular traps (NETs), neutrophils help limit viral spread during the acute phase of infection (77). Neutrophils also produce cytokines and extracellular matrix-degrading proteases, which cause inflammation and tissue destruction, often leading to blindness (18). Most of the damage is done through neutrophil infiltration and neovascularization since neutrophils release cytokines and chemokines, which are proinflammatory agents; however, there are some cytokines and chemokines that are anti-inflammatory agents, which could be further studied and used in future therapies (78).
4.5 Mast cells
MCs function as effector, initiator, and regulator cells in innate immune responses, acting as important sentinels against infection by releasing a diverse array of inflammatory molecules such as cytokines and chemokines (79). MCs typically operate through TLR signaling, using TLR3, TLR7, and TLR9 induced activation to initiate production of an inflammatory response to virus related PAMPs (80). A previous study has shown evidence of MCs contribution to protection against HSV-2, using a “MC knock-in” mouse model to show increased production of TNF-α and IL-6 following skin infection by HSV-2 (81). However, the contribution of MCs to both ocular infection and infection by HSV-1 still requires further evidence to establish a potential relationship between MCs and HSK.
5 Role of sensors in DNA sensing signaling pathway in HSK
Innate immunity is the first line of defense against DNA virus HSV-1. Activation of innate immunity usually requires the recognition of viral PAMPs, such as dsDNA from HSV-1, by PRRs on innate immune cells (19, 29, 82). However, DNA sensors can also recognize endogenous DNA released during cellular damage or stress, triggering immune responses that clear damaged cells and induce cytokines release (83).The cytoplasmic DNA sensors involved in HSV-1 detection include cGAS (20), interferon gamma-inducible protein 16 (IFI16) (84), DEAD-box helicase 41 (DDX41) (85), and absent in melanoma 2 (AIM2) (86), which recognize double-stranded DNA in the cytoplasm and trigger the production of type I IFN through stimulator of interferon genes (STING) signaling (29, 30, 87). The roles of these DNA sensors in HSV-1 recognition are discussed below.
5.1 cGAS
The cGAS-STING pathway plays an important role in host antiviral immune responses and its interactions with viral immune escape mechanisms are very important for limiting HSV-1 lysis and latent infection (88, 89). When HSV-1 virus is being replicated, the cGAS enzyme senses aberrant DNA and catalyzes the cyclic cGAMP to activate STING receptors inside the cell (90, 91). Recognition of this process activates the interferon regulatory factor 3 (IRF3) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathways, which in turn promotes the secretion of type I IFN and other pro-inflammatory cytokines (92). Beta-conjugated proteins can also promote type I IFN production in the cGAS-cGAMP-STING pathway, which can better apply anti-HSV-1 effects (93) (Figure 2).
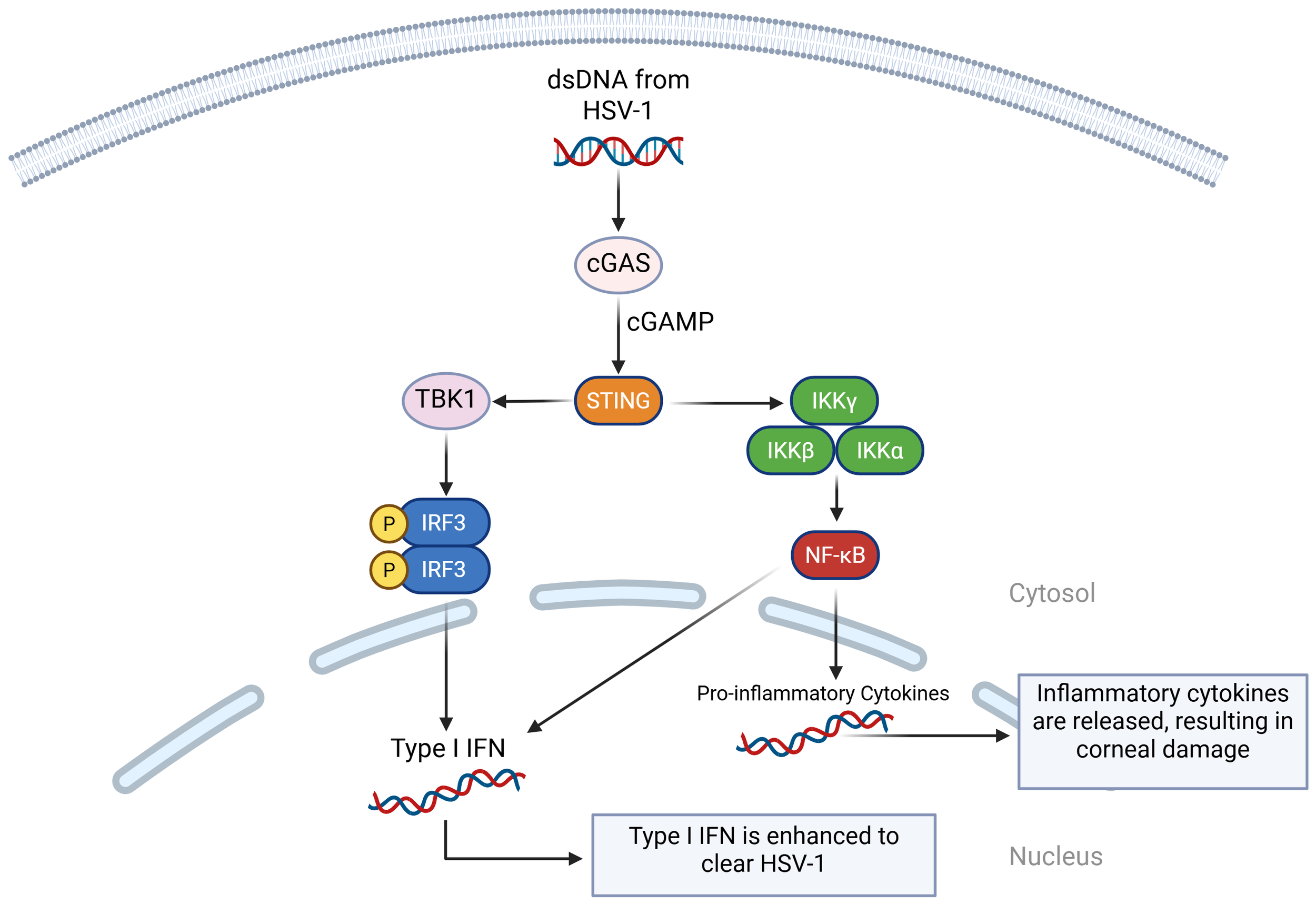
A visual diagram of the cGAS-cGAMP-STING pathway in HSV-1 infection. When cGAS detects double-stranded DNA (dsDNA) from HSV-1, it binds to it and activates enzymatic activity. This catalyzes the formation of cGAMP, which binds it to STING. STING recruits TANK-binding kinase 1 (TBK1), which phosphorylates IRF3, activating type I IFN. STING also activates IκB kinase (IKK) complex, which activates NF-κB. NF-κB translocates to the nucleus and initiates the transcription of inflammatory genes. It also works with IRF3 to initiate type I IFN.
5.2 IFI16
IFI16 has an important role in antiviral defense by activating the canonical STING/TANK binding kinase 1 (TBK1)/IRF3 signaling pathway in response to viral infections (87). During the HSV-1 infection, IFI16 recognizes and binds dsDNA in the nucleus, blocking the virus’s ability to turn its genes into proteins (94). This then leads to the production of interferons and many antiviral proteins, such as mucosal viral resistance (MxA, a GTPase), 2′-5′-oligoadenylate synthetase (OAS), and ribonuclease L (RNase L) (95). All of this stops the virus from spreading. Additionally, if IFI16 is absent, then expression of type I IFN and type III IFN is significantly reduced (96). Ubiquitin-specific peptidase 12 (USP12) promotes antiviral responses by removing ubiquitin molecules from proteins and stabilizing IFI16 (97). Like the duality of TLR2/TLR9, cGAS and IFI16 can co-recognize HSV-1 and stimulate the IRF3 pathway while also restricting viral replication by binding to viral genomes and activating inflammasomes, which are an essential part of the immune system response (98) (Figure 3).
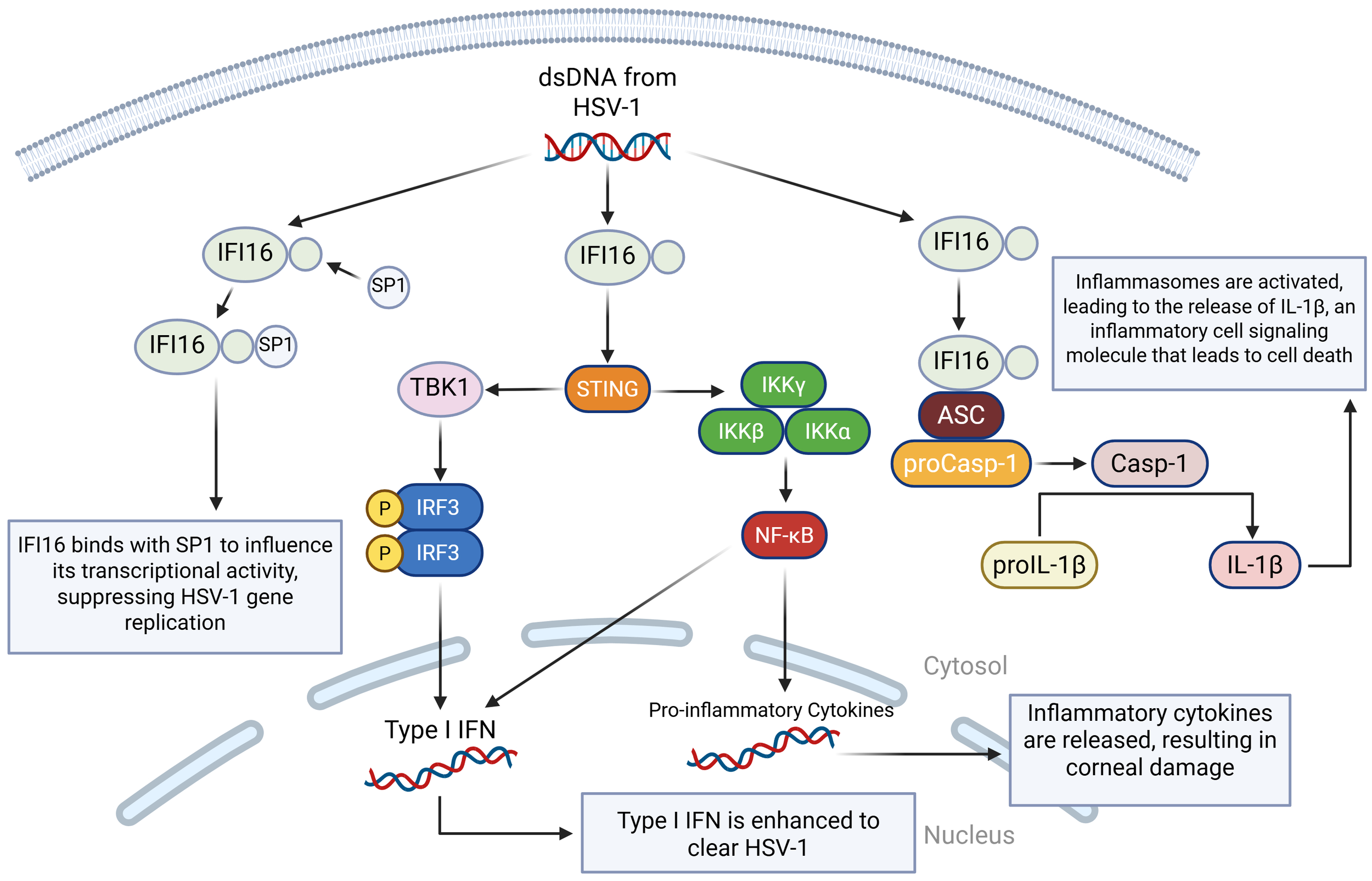
A visual diagram of the IFI16 DNA sensing pathways in HSV-1 infection. IFI16 binds to dsDNA from HSV-1. In the first pathway, IFI16 binds to the specificity protein 1 (SP1), modulating SP1’s ability to regulate antiviral genes, which ultimately leads to the restriction of HSV-1 viral replication by inducing an antiviral state. In the second pathway, the STING-TBK1-IRF3 pathway is activated, leading to type I and type III IFN production. In the third pathway, IFI16 binds to the apoptosis-associated speck-like protein (ASC), which binds to procaspase 1 (proCasp-1), facilitating the activation of Casp-1 and completing the activation of the inflammasome. Casp-1 then cleaves pro-interleukin-1 beta (proIL-1β) so that it can become mature IL-1β, which is a highly inflammatory cell signaling molecule that leads to pyroptotic cell death.
5.3 DDX41
DDX41 is an intracellular DNA sensor that triggers the downstream pathway, requiring the adaptor STING, the kinase TBK1, and the transcription factor IRF3 to activate the type I IFN response (85), and it also plays an important role in modulating dsDNA and ssDNA from HSV-1 while also activating the DDX41– Receptor-interacting protein kinase 3 – Mixed lineage kinase domain-like protein (DDX41-RIPK3-MLKL), which results in necroptosis (99). A study was performed to screen, identify, and characterize HSV-1-encoded microRNA H2-3p (miR-H2-3p) as a suppressor of the cytosolic DNA-stimulated antiviral innate immune pathway by targeting DNA sensor DDX41 to neutralize the production of type I IFN and strengthen HSV-1 immune evasion (100) (Figure 4).
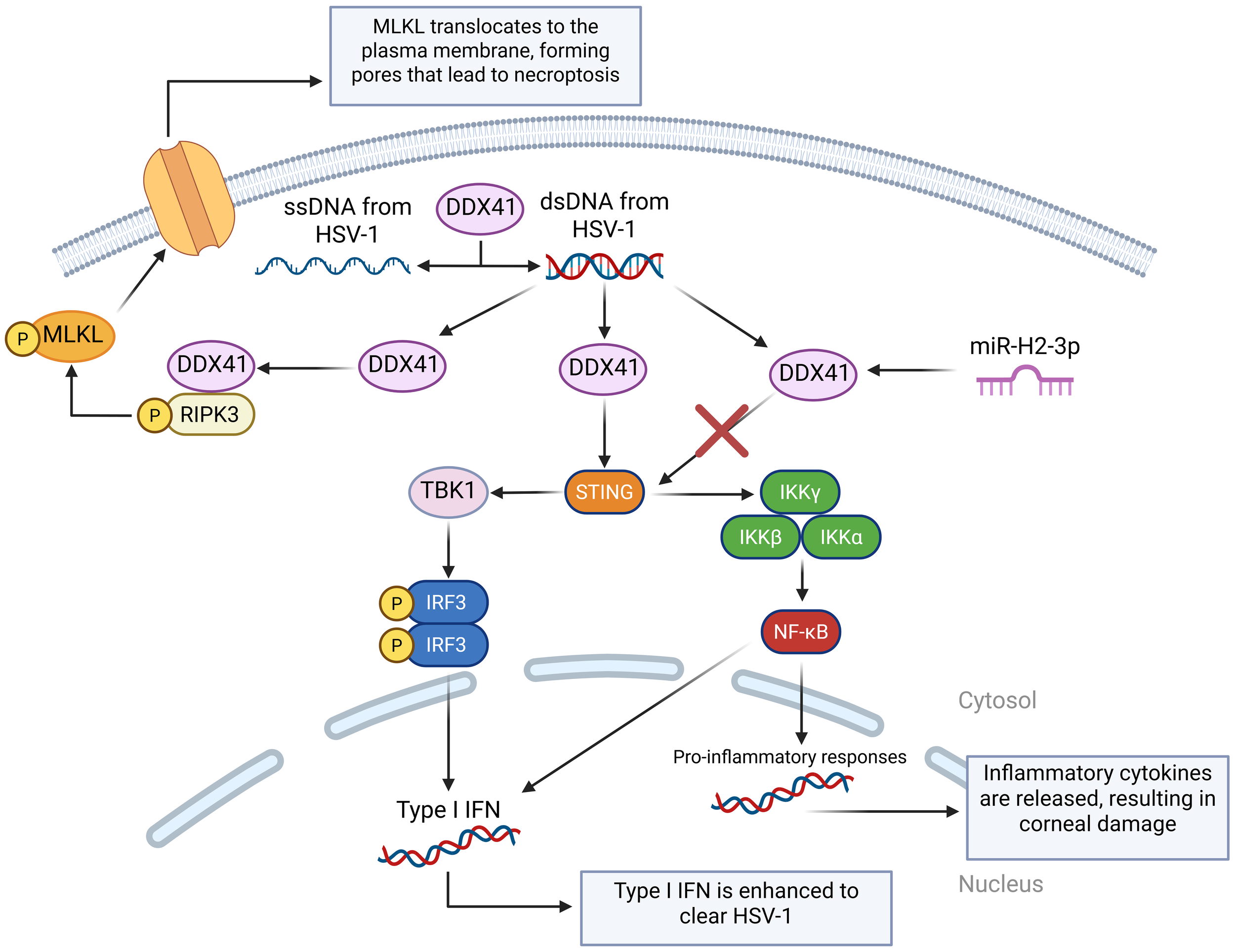
A visual diagram of the DDX41 DNA sensing pathways in HSV-1 infection. DDX41 modulates the state of cytosolic DNA by unwinding dsDNA and annealing ssDNA. This is very important in regulating cGAS activation. Like cGAS, DDX41 can also activate the STING-TBK-IRF3 pathway. miR-H2-3p targets DDX41, preventing it from activating the STING pathway and reducing the cell’s immune response. DDX41 also binds with Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3) to activate the Mixed Lineage Kinase Domain-Like protein (MLKL), which translocates to the cell membrane and forms pores, disrupting the cell’s ion balance to cause necroptosis.
5.4 AIM2
AIM2 is a DNA sensor that detects foreign dsDNA in the cytoplasm, which comes from viruses like cytomegalovirus (CMV) and HSV-1 (101). When AIM2 detects dsDNA from HSV-1, it assembles an inflammasome, which is a multi-protein complex that forms inside the cell as part of the innate immune system (102). The role of inflammasomes is to detect dangerous signals from foreign invaders and trigger proptosis, a form of programmed cell death (103). The activation of the AIM2 inflammasome is triggered by dsDNA, which then results in the activation of caspase-1 and the release of pro-inflammatory cytokines IL-1β and IL-18, which play an important role in the inflammatory response of cells (86). Investigation into the AIM2 inflammasome unveiled that HSV-1 triggered the activation of AIM2 in macrophages independently of the dsDNA sensor, which means that HSV-1 can activate AIM2 without relying on the usual DNA-sensing mechanism (104). HSV-1 tegument protein VP22 (VP22), was identified as a specific inhibitor of the AIM2 inflammasome during HSV-1 infection, meaning that HSV-1 tries to block AIM2 using the protein VP22 to inhibit detection (105) (Figure 5).
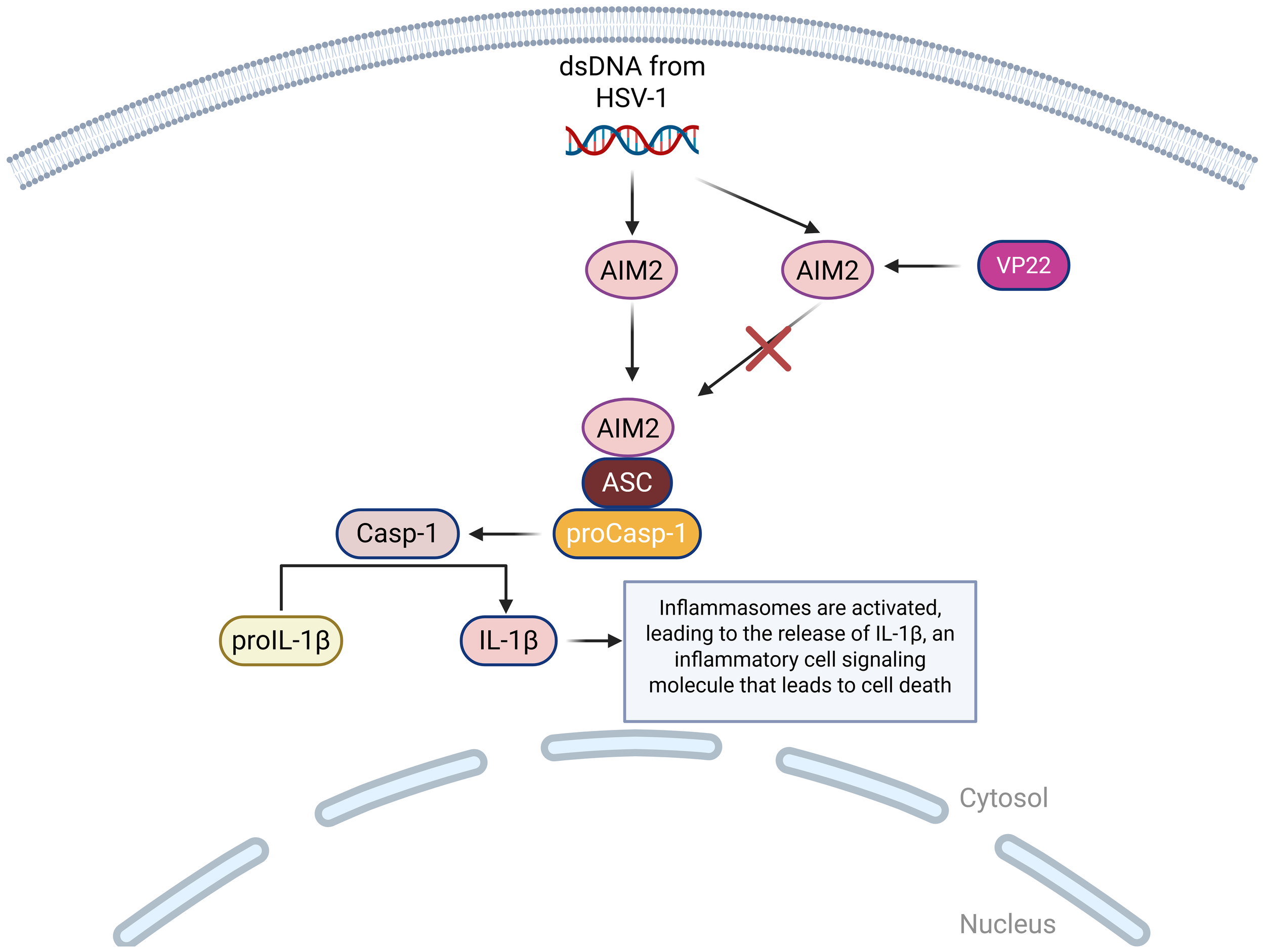
A visual diagram of the AIM2 DNA sensing pathways in HSV-1 infection. AIM2 binds to ASC, which binds to proCasp-1, facilitating the activation of Casp-1 and completing the activation of the inflammasome. Casp-1 then cleaves proIL-1β so that it can become mature IL-1β, which is a highly inflammatory cell signaling molecule that leads to pyroptotic cell death. This pathway can be inhibited by HSV-1 encoded protein VP22, which suppresses the AIM2 inflammasome activation.
6 Role of regulators in DNA sensing signaling pathway in HSK6.1 TRIM family proteins
TRIM proteins, including 80 members in humans, are E3 ubiquitin ligases and play extremely important roles in regulating innate immune sensing, interferon production, and antiviral restriction (106, 107). TRIM21 can play a significant role in HSK (108). TRIM21 regulates the type I IFN response to viruses (109), and also serves as a cytosolic Fc receptor for immunoglobulin (110). HSV-1 is sensitive to type I IFN and neutralizing antibody, and the role of TRIM21 in the response to ocular HSV-1 infection in mice has been investigated (111). It has been shown that the absence of TRIM21 results in a significant increase in HSV-1 titers recovered from the thapsigargin (TG) of TRIM21 KO mice during HSV-1 infection (112). In epithelial HSK mice models, the expression TRIM21 was detected, and the clinical relationship was then investigated between TRIM21 and epithelial HSK in which TRIM21 was silenced, significantly controlling viral particle release at 1, 3, and 5 days post-HSV-1 infection (113). Ultimately, clinical scores and histopathology examinations have shown that TRIM21 can successfully reduce the severity of epithelial HSK (114).
TRIM29 has been shown to play important roles in host defense against both DNA and RNA viruses through regulating host innate immune responses mediated by type I IFN, IFN-γ, and inflammasomes (49, 69, 115–118). Specifically, TRIM29 interacts with STING to induce K48-linked ubiquitination and degradation of STING, thereby reducing type I IFN production in DCs, leading to increased HSV-1 replication and pathogenesis in vivo (116). Our unpublished data shows that TRIM29 is highly expressed in CECs, suggesting that TRIM29 plays a key role in controlling HSV-1 infection and may influence the severity of HSK.
TRIM18 is an E3 ubiquitin ligase that plays a negative regulatory role in the innate immune response to both DNA and RNA viruses. TRIM18 is shown to recruit protein phosphatase 1A (PPM1A) to dephosphorylate TBK1, which deactivates TBK1 to block TBK1 from interacting with its upstream adaptor STING in macrophages, thereby dampening type I IFN-mediated antiviral signaling during HSV-1 infection (62). Given that the critical role of macrophages in regulating HSK, we hypothesize that TRIM18 could regulate antiviral innate immunity in macrophages to control HSK.
6.2 TLR2
TLR2 is shown to detect viral glycoproteins, including HSV-1 glycoproteins gB and gH, signaling through MyD88 to activate NF-κB and mitogen-activated protein kinase (MAPK) pathways, leading to pro-inflammatory cytokine production (76). While not a DNA sensor itself, TLR2 can indirectly regulate DNA sensing pathways, such as cGAS-STING, through inflammatory priming and signaling crosstalk (119). For example, TLR2-induced cytokines like IL-1β can enhance STING pathway activation, thereby regulating type I IFN production downstream of DNA sensors (120). In murine models, TLR2 is critical for early innate responses in the cornea, with TLR2-deficient mice showing lower early inflammatory cytokine levels, reduced recruitment of neutrophils and monocytes, and decreased severity of corneal immunopathology (121). Therefore, while TLR2 helps detect HSV early, excessive TLR2 signaling drives corneal opacity, neovascularization, and scarring, ultimately damaging the cornea and causing the progression of HSK (122).
6.3 NLRP3
NLRP3 is a cytosolic PRR that forms the NLRP3 inflammasome, and upon activation, NLRP3 recruits apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1, driving inflammation (123). HSV-1 infection triggers NLRP3 inflammasome activation in corneal epithelial cells, stromal keratocytes, and infiltrating leukocytes (124). NLRP3-deficient mice infected with HSV-1 show reduced IL-1β secretion, lower neutrophil infiltration, and less corneal opacity and neovascularization. However, viral titers can remain similar, indicating that NLRP3 mainly drives immunopathology rather than clearance (125).
7 HSV-1 evasion of host innate immunity
Although the cytosolic DNA sensing signaling pathway is activated during viral infection, HSV-1 has developed multiple mechanisms to evade host antiviral innate immunity and to facilitate viral infection and replication (22, 89, 126). HSV-1 encoded proteins US11 (127), US3 (128), UL36 (129), and VP16 (130) can evade RNA sensing antiviral signaling pathways, while UL41 (131), VP24 (132), ICP0 (133), and ICP27 (134) proteins evade DNA sensing antiviral signaling pathways. Additionally, the tegument protein VP22 inhibits AIM2-dependent inflammasome responses (102). HSV-1 can also block autophagy in order to evade innate immunity. It accomplishes this through ICP34.5, which binds to Beclin-1, a key autophagy protein (135). Finally, HSV-1 can interfere with NK cells activation signals by downregulating ligands that bind to the NK-cell activating receptor NKG2D, which limits NK cells recognition and cytotoxic killing (136).
8 Therapeutic treatments for HSK8.1 Antiviral drug therapies
Antiviral drugs, including acyclovir, trifluridine, ganciclovir, vidarabine, and famciclovir (137), are commonly used to treat HSK. Oral acyclovir, when added to primary treatment with topical corticosteroids and trifluridine, does not significantly improve initial outcomes but may provide long-term vision benefits (138). Trifluridine, a nucleoside analog, inhibits viral DNA synthesis, preventing HSV-1 replication. Higher doses have been shown to reduce the risk of antiviral resistance (139). In epithelial HSK, topical trifluridine or ganciclovir is standard, with optional oral acyclovir. For stromal and endothelial HSK, oral acyclovir is combined with topical corticosteroids (140).
8.2 Host-directed therapies
Host-directed therapies (HDTs) enhance host immune responses by targeting host factors critical for viral pathogenesis, offering a promising alternative to conventional antivirals for HSK (141). Unlike traditional antivirals, HDTs focus on host pathways to disrupt viral entry, replication, or immune evasion, potentially overcoming resistance to drugs like acyclovir. HSV-1 entry begins with glycoproteins binding to host cell receptors, such as heparan sulfate proteoglycans (HSPGs), followed by interactions with key glycoprotein D (gD) receptors: herpesvirus entry mediator (HVEM), nectin-1, and 3-O-sulfated heparan sulfate (3-OS HS). These interactions activate gB, facilitating membrane fusion and viral entry. HDTs can block or modify these attachment sites, particularly 3-OS HS, or use inhibitors, antagonists, or decoy molecules to disrupt HVEM or nectin-1 binding, preventing viral entry (142). HSV-1 infection also upregulates host kinases, which serve as viable therapeutic targets. For instance, the cyclin-dependent kinase (CDK) inhibitor FIT-039 disrupts mRNA transcription, inhibiting replication of various DNA viruses, including HSV-1, as shown in animal models (143). Similarly, BX795 hydrochloride, a serine/threonine kinase inhibitor, blocks viral protein synthesis, demonstrating efficacy against acyclovir-resistant HSV-1 strains in a mouse HSK model (144). Additionally, HDTs can amplify antiviral immunity by targeting immune cells. For example, overexpression of NLRP12 enhances macrophage immune responses to alleviate HSK (63), and targeted delivery of HSV-1 gD to CD169+ macrophages using ganglioside liposomes reduces HSK severity in mice (10). DCs are also critical targets, as local cDCs depletion results in decreased corneal nerve infection and mortality of mice (145), while pDCs depletion leads to severe HSK (37). Modulating DCs function could thus enhance antiviral defenses and mitigate HSK progression (146). Overall, HDTs offer a multifaceted approach to HSK treatment by targeting viral entry, host kinases, and immune cell responses, providing potential solutions for drug-resistant strains and improving therapeutic outcomes.
8.3 Anti-inflammatory therapies
Corticosteroids are anti-inflammatory and immunosuppressive drugs that decrease the production of inflammatory cytokines by binding to glucocorticoid receptors inside cells, and they are important in treating HSK because they can prevent inflammatory complications, slowing down vision impairment (147). Some corticosteroids include prednisolone acetate and dexamethasone, and they are always used with antivirals to prevent uncontrolled HSV-1 replication (148). Another type of anti-inflammatory therapy is non-steroidal anti-inflammatory drugs (NSAIDs), which are a type of medication that block cyclooxygenase enzymes in order to reduce inflammation (137). Topical NSAIDs, such as flurbiprofen and diclofenac, inhibit prostaglandin synthesis, thereby decreasing vasodilation, vascular permeability, and pain signaling (149).
8.4 Novel gene therapies
One of the most promising novel gene therapies is HSV-1-erasing lentiviral particles (HELP). This gene therapy uses virus-like particles to deliver SpCas9 mRNA and single-guide RNAs (sgRNAs) targeting HSV-1 genes UL8 and UL29 via corneal intrastromal injection. Preclinical studies demonstrate complete inhibition of HSV-1 replication and prevention of HSK in multiple animal models (2). Another gene therapy is meganuclease-based gene editing, which uses a meganuclease to target HSV-1 UL19, and it is delivered via adeno-associated virus serotype 2 (AAV2) to corneal grafts. In rabbit models, treated corneal transplants resisted HSV-1 infection, preventing opacity and edema (150). Host gene targeting is another type of gene therapy that uses CRISPR to edit NECTIN-1, an essential HSV-1 entry receptor on CECs. Studies have shown that lentiviral delivery in vitro dramatically lowered infection rates and viral load (151).
8.5 Combination therapies
Combination therapies address both viral replication and immune-mediated corneal damage in HSK management. The most common type of combination therapy is antivirals and corticosteroids. This is especially effective in treating stromal and endothelial HSK, and an example would be oral acyclovir combined with topical prednisolone acetate (152). Another type of combination therapy is antivirals and NSAIDS, which is typically used when steroids are contraindicated. An example is topical trifluridine and topical flurbiprofen, but the downside to this combination treatment is that NSAIDs are less effective than corticosteroids for stromal inflammation (153). Finally, antivirals can be combined with surgical interventions, which is a treatment method that is used in severe recurrent HSK with scarring. An example is oral acyclovir prophylaxis and penetrating keratoplasty, which is a treatment plan that has been shown to reduce the risk of HSK recurrence and graft failure (154) (Figure 6).
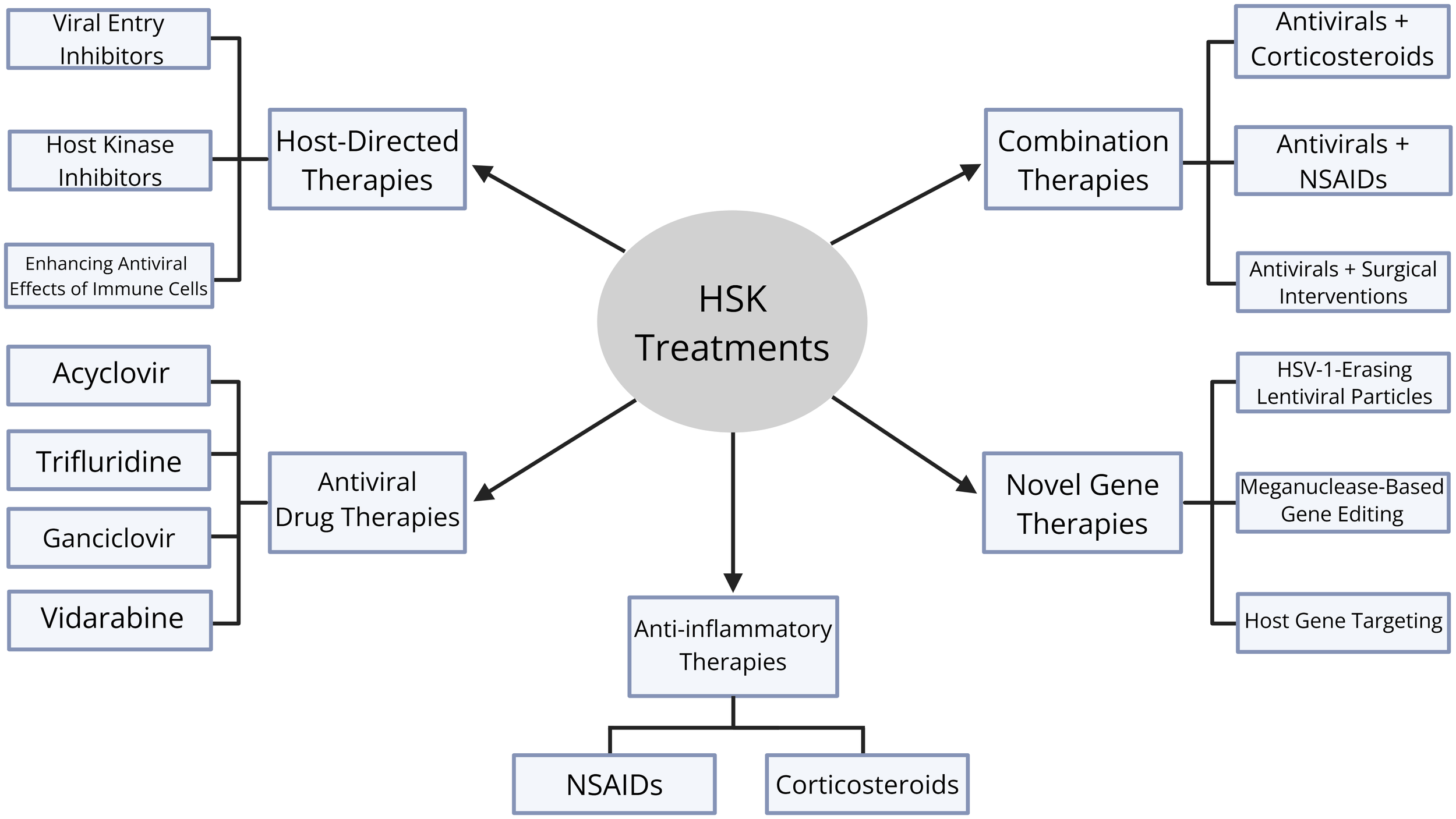
Diagram of therapeutic treatments for HSK. There are five categories of therapeutic treatments: antiviral drug therapies, host-directed therapies, anti-inflammatory therapies, novel gene therapies, and combination therapies. Antiviral drug therapies include acyclovir, trifluridine, ganciclovir, and vidarbine. Host-directed therapies include viral entry inhibitors, host kinase inhibitors, and enhancing antiviral effects of immune cells. Anti-inflammatory therapies include corticosteroids and non-steroidal anti-inflammatory drugs (NSAIDs). Novel gene therapies include HSV-1-erasing lentiviral particles, meganuclease-based gene editing, and host gene targeting. Finally, combination therapies include antivirals and corticosteroids, antivirals and NSAIDs, and antivirals and surgical interventions.
9 Conclusions and future perspectives
HSK is a complex disease that involves many different aspects, including the viral infection itself, the immune system’s reaction, molecular regulation, and inflammation. HSK is the most common cause of infectious blindness, and the current methods of treatment are unsatisfactory. As a dsDNA virus that exhibits a strong neurotropic nature, treatment can be extremely difficult since it persists in neuronal tissues and can exist in a latent phase while preventing host immune clearance. With many different DNA sensing pathways, the immune system’s response to HSV-1 is extremely complex and involves many interconnected interactions between various immune cells. Novel insights into disease immunopathogenesis could allow for the development of more efficient and effective therapeutic options. Current therapies, while effective at controlling viral replication, are limited in preventing corneal scarring, opacity, and neovascularization. Because of this, increasing attention has turned toward host-directed therapies that modulate innate immune responses. Some potential targets for future host-directed therapies include cGAS-STING, NLRP3, IL-17, and TRIM proteins. Looking forward, integrating antiviral agents with precision immunomodulation offers a path forward for more effective and personalized HSK management. Future research should prioritize clinical translation of host-targeted interventions as well as combination strategies that balance viral control with immune regulation.
StatementsAuthor contributions
PN: Writing – original draft. BJ: Writing – original draft. AM: Writing – original draft. CH: Writing – original draft. JX: Project administration, Conceptualization, Validation, Funding acquisition, Writing – review & editing, Supervision.
Funding
The author(s) declare financial support was received for the research and/or publication of this article. This work was supported by the American Heart Association Career Development Award 20CDA35260116 and Transformational Project Award 23TPA1055437 (https://doi.org/10.58275/AHA.23TPA1055437.pc.gr.172259) (JX).
Acknowledgments
We thank Dr. Xian C. Li and Dr. Zhiqiang Zhang (Houston Methodist) for their excellent editing and advice on this review manuscript. We apologize to all colleagues whose contributions were not discussed and cited owing to space constraints. Figures were created with BioRender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Any alternative text (alt text) provided alongside figures in this article has been generated by Frontiers with the support of artificial intelligence and reasonable efforts have been made to ensure accuracy, including review by the authors wherever possible. If you identify any issues, please contact us.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
AntonyFKinhaDNowińskaARouseBTSuryawanshiA. The immunobiology of corneal HSV-1 infection and herpetic stromal keratitis. Clin Microbiol Rev. (2024) 37:e0000624. doi: 10.1128/cmr.00006-24
YinDLingSWangDDaiYJiangHZhouXet al. Targeting herpes simplex virus with CRISPR-Cas9 cures herpetic stromal keratitis in mice. Nat Biotechnol. (2021) 39:567–77. doi: 10.1038/s41587-020-00781-8
WhitleyRJRoizmanB. Herpes simplex virus infections. Lancet. (2001) 357:1513–8. doi: 10.1016/S0140-6736(00)04638-9
McCormickIJamesCWeltonNJMayaudPTurnerKMEGottliebSLet al. Incidence of herpes simplex virus keratitis and other ocular disease: global review and estimates. Ophthalmic Epidemiol. (2022) 29:353–62. doi: 10.1080/09286586.2021.1962919
ShenYZhangHXueMZhengCChenQ. HSV-1 as a gene delivery platform for cancer gene therapy. Trends Pharmacol Sci. (2025) 46:324–36. doi: 10.1016/j.tips.2025.02.006
BearerELBreakefieldXOSchubackDReeseTSLaVailJH. Retrograde axonal transport of herpes simplex virus: evidence for a single mechanism and a role for tegument. Proc Natl Acad Sci U.S.A. (2000) 97:8146–50. doi: 10.1073/pnas.97.14.8146
NicollMPHannWShivkumarMHarmanLEConnorVColemanHMet al. The HSV-1 latency-associated transcript functions to repress latent phase lytic gene expression and suppress virus reactivation from latently infected neurons. PLoS Pathog. (2016) 12:e1005539. doi: 10.1371/journal.ppat.1005539
KapoorDSharmaPShuklaD. Emerging drugs for the treatment of herpetic keratitis. Expert Opin Emerg Drugs. (2024) 29:113–26. doi: 10.1080/14728214.2024.2339899
LiLLiYLiXXiaYWangEGongDet al. HSV-1 infection and pathogenesis in the tree shrew eye following corneal inoculation. J Neurovirol. (2020) 26:391–403. doi: 10.1007/s13365-020-00837-0
ShenWWangCJiangJHeYLiangQHuK. Targeted delivery of herpes simplex virus glycoprotein D to CD169+ macrophages using ganglioside liposomes alleviates herpes simplex keratitis in mice. J Control Release. (2024) 365:208–18. doi: 10.1016/j.jconrel.2023.11.026
SuryawanshiRKPatilCDAgelidisAKogantiRAmesJMKoujahLet al. mTORC2 confers neuroprotection and potentiates immunity during virus infection. Nat Commun. (2021) 12:6020. doi: 10.1038/s41467-021-26260-5
SuryawanshiRKPatilCDBoraseHShuklaD. Akt isoform specificity drives intrinsic immune regulation during HSV-1 infection. Proc Natl Acad Sci U.S.A. (2025) 122:e2504962122. doi: 10.1073/pnas.2504962122
GarciaLSde SousaRMPCamposVSFerreiraEMCascabulhoCMde SouzaEMet al. CRISPR/cas9 reduces viral load in a BALB/c mouse model of ocular herpes infection. Biomedicines. (2025) 13:1738. doi: 10.3390/biomedicines13071738
ChewTTaylorKEMossmanKL. Innate and adaptive immune responses to herpes simplex virus. Viruses. (2009) 1:979–1002. doi: 10.3390/v1030979
CookSDHillJMLynasCMaitlandNJ. Latency-associated transcripts in corneas and ganglia of HSV-1 infected rabbits. Br J Ophthalmol. (1991) 75:644–8. doi: 10.1136/bjo.75.11.644
YadavalliTPatilCSharmaPVoletyIBoraseHKapoorDet al. Unique attributes of Guinea pigs as new models to study ocular herpes pathophysiology and recurrence. Invest Ophthalmol Vis Sci. (2023) 64:41. doi: 10.1167/iovs.64.14.41
JamesCHarfoucheMWeltonNJTurnerKMAbu-RaddadLJGottliebSLet al. Herpes simplex virus: global infection prevalence and incidence estimates, 2016. Bull World Health Organ. (2020) 98:315–29. doi: 10.2471/BLT.19.237149
LoboAMAgelidisAMShuklaD. Pathogenesis of herpes simplex keratitis: The host cell response and ocular surface sequelae to infection and inflammation. Ocul Surf. (2019) 17:40–9. doi: 10.1016/j.jtos.2018.10.002
TakeuchiOAkiraS. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
SunLWuJDuFChenXChenZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. (2013) 339:786–91. doi: 10.1126/science.1232458
TakeuchiOAkiraS. Innate immunity to virus infection. Immunol Rev. (2009) 227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x
ZhuHZhengC. The race between host antiviral innate immunity and the immune evasion strategies of herpes simplex virus 1. Microbiol Mol Biol Rev. (2020) 84:e00099-20. doi: 10.1128/MMBR.00099-20
AzherTNYinXTTajfirouzDHuangAJStuartPM. Herpes simplex keratitis: challenges in diagnosis and clinical management. Clin Ophthalmol. (2017) 11:185–91. doi: 10.2147/OPTH.S80475
SibleyDLarkinDFP. Update on Herpes simplex keratitis management. Eye (Lond). (2020) 34:2219–26. doi: 10.1038/s41433-020-01153-x
MoeinHRKheirkhahAMullerRTCruzatACPavan-LangstonDHamrahP. Corneal nerve regeneration after herpes simplex keratitis: A longitudinal in vivo confocal microscopy study. Ocul Surf. (2018) 16:218–25. doi: 10.1016/j.jtos.2017.12.001
ValerioGSLinCC. Ocular manifestations of herpes simplex virus. Curr Opin Ophthalmol. (2019) 30:525–31. doi: 10.1097/ICU.0000000000000618
SwaroopR. Conjunctival geographic ulcer and blepharitis in primary ocular herpes: a case report. cases J. (2009) 2:8141. doi: 10.4076/1757-1626-2-8141
CarrDJTomanekL. Herpes simplex virus and the chemokines that mediate the inflammation. Curr Top Microbiol Immunol. (2006) 303:47–65. doi: 10.1007/978-3-540-33397-5_3
LinRXingJZhengC. Editorial: sensing DNA in antiviral innate immunity. Front Immunol. (2021) 12:644310. doi: 10.3389/fimmu.2021.644310
LuWWangLXingJ. Editorial: Antiviral innate immune sensing, regulation, and viral immune evasion. Front Immunol. (2023) 14:1358542. doi: 10.3389/fimmu.2023.1358542
WangLXingJ. Editorial: Community series in antiviral innate immune sensing, regulation, and viral immune evasion: volume II. Front Immunol. (2023) 14:1341193. doi: 10.3389/fimmu.2023.1341193
HungSLChiangHHWuCYHsuMJChenYT. Effects of herpes simplex virus type 1 infection on immune functions of human neutrophils. J Periodontal Res. (2012) 47:635–44. doi: 10.1111/j.1600-0765.2012.01476.x
KnickelbeinJEHendricksRLCharukamnoetkanokP. Management of herpes simplex virus stromal keratitis: an evidence-based review. Surv Ophthalmol. (2009) 54:226–34. doi: 10.1016/j.survophthal.2008.12.004
NewellCKMartinSSendeleDMercadalCMRouseBT. Herpes simplex virus-induced stromal keratitis: role of T-lymphocyte subsets in immunopathology. J Virol. (1989) 63:769–75. doi: 10.1128/jvi.63.2.769-775.1989
InoueY. Review of clinical and basic approaches to corneal endotheliitis. Cornea. (2014) 33 Suppl:11, S3–8. doi: 10.1097/ICO.0000000000000228
GiménezFSuryawanshiARouseBT. Pathogenesis of herpes stromal keratitis–a focus on corneal neovascularization. Prog Retin Eye Res. (2013) 33:1–9. doi: 10.1016/j.preteyeres.2012.07.002
JamaliAHuKSendraVGBlancoTLopezMJOrtizGet al. Characterization of resident corneal plasmacytoid dendritic cells and their pivotal role in herpes simplex keratitis. Cell Rep. (2020) 32:108099. doi: 10.1016/j.celrep.2020.108099
SmithJBHerbertJJTruongNRCunninghamAL. Cytokines and chemokines: The vital role they play in herpes simplex virus mucosal immunology. Front Immunol. (2022) 13:936235. doi: 10.3389/fimmu.2022.936235
McKayTBHutcheonAEKZieskeJDCiolinoJB. Extracellular vesicles secreted by corneal epithelial cells promote myofibroblast differentiation. Cells. (2020) 9:1080. doi: 10.3390/cells9051080
SteppMAMenkoAS. Immune responses to injury and their links to eye disease. Transl Res. (2021) 236:52–71. doi: 10.1016/j.trsl.2021.05.005
VendomèleJKhebiziQFissonS. Cellular and molecular mechanisms of anterior chamber-associated immune deviation (ACAID): what we have learned from knockout mice. Front Immunol. (2017) 8:1686. doi: 10.3389/fimmu.2017.01686
NiederkornJY. Role of NKT cells in anterior chamber-associated immune deviation. Expert Rev Clin Immunol. (2009) 5:137–44. doi: 10.1586/1744666X.5.2.137
Gonzalez-DosalRHoranKARahbekSHIchijoHChenZJMieyalJJet al. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: role for S-glutathionylation of TRAF3 and 6. PLoS Pathog. (2011) 7:e1002250. doi: 10.1371/journal.ppat.1002250
ChangJYaoYSunXWangWQianHLiuYet al. JAG1 mediates apoptosis in herpes simplex keratitis by suppressing autophagy via ROS/JAG1/NOTCH1/pULK1 signaling pathway. Cell Biol Toxicol. (2024) 41:1. doi: 10.1007/s10565-024-09968-0
RenJAntonyFRouseBTSuryawanshiA. Role of innate interferon responses at the ocular surface in herpes simplex virus-1-induced herpetic stromal keratitis. Pathogens. (2023) 12:437. doi: 10.3390/pathogens12030437
Ayilam RamachandranRLemoffARobertsonDM. Extracellular vesicles released by host epithelial cells during Pseudomonas aeruginosa infection function as homing beacons for neutrophils. Cell Commun Signal. (2024) 22:341. doi: 10.1186/s12964-024-01609-7
JiangJShenWHeYLiuJOuyangJZhangCet al. Overexpression of NLRP12 enhances antiviral immunity and alleviates herpes simplex keratitis via pyroptosis/IL-18/IFN-γ signaling. Int Immunopharmacol. (2024) 137:112428. doi: 10.1016/j.intimp.2024.112428
RoneyK. Bone marrow-derived dendritic cells. Methods Mol Biol. (2019) 1960:57–62. doi: 10.1007/978-1-4939-9167-9_4
XingJZhangAMinzeLJLiXCZhangZ. TRIM29 negatively regulates the type I IFN production in response to RNA virus. J Immunol. (2018) 201:183–92. doi: 10.4049/jimmunol.1701569
XingJZhangADuYFangMMinzeLJLiuYJet al. Identification of poly(ADP-ribose) polymerase 9 (PARP9) as a noncanonical sensor for RNA virus in dendritic cells. Nat Commun. (2021) 12:2681. doi: 10.1038/s41467-021-23003-4
KwonMSCarntNATruongNRPattamattaUWhiteAJSamarawickramaCet al. Dendritic cells in the cornea during Herpes simplex viral infection and inflammation. Surv Ophthalmol. (2018) 63:565–78. doi: 10.1016/j.survophthal.2017.11.001
HamrahPPavan-LangstonDDanaR. Herpes simplex keratitis and dendritic cells at the crossroads: lessons from the past and a view into the future. Int Ophthalmol Clin. (2009) 49:53–62. doi: 10.1097/IIO.0b013e3181924dd8
FrankGMBuelaKAMakerDMHarveySAHendricksRL. Early responding dendritic cells direct the local NK response to control herpes simplex virus 1 infection within the cornea. J Immunol. (2012) 188:1350–9. doi: 10.4049/jimmunol.1101968
SimWJMalinarichFFairhurstAMConnollyJE. Generation of immature, mature and tolerogenic dendritic cells with differing metabolic phenotypes. J Vis Exp. (2016) 112:54128. doi: 10.3791/54128
Shapouri-MoghaddamAMohammadianSVaziniHTaghadosiMEsmaeiliSAMardaniFet al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. (2018) 233:6425–40. doi: 10.1002/jcp.26429
ZhangJXingJ. Editorial: Plasticity and metabolic switching in adipose tissue macrophages. Front Immunol. (2023) 14:1233791. doi: 10.3389/fimmu.2023.1233791
LeeDHJaggiUGhiasiH. CCR2+ migratory macrophages with M1 status are the early-responders in the cornea of HSV-1 infected mice. PLoS One. (2019) 14:e0215727. doi: 10.1371/journal.pone.0215727
MartinezFOGordonS. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. (2014) 6:13. doi: 10.12703/P6-13
FairweatherDCihakovaD. Alternatively activated macrophages in infection and autoimmunity. J Autoimmun. (2009) 33:222–30. doi: 10.1016/j.jaut.2009.09.012
ZyzakJMitkiewiczMLeszczyńskaEReniewiczPMoynaghPNSiednienkoJ. HSV-1/TLR9-mediated IFNβ and TNFα Induction is mal-dependent in macrophages. J Innate Immun. (2020) 12:387–98. doi: 10.1159/000504542
NiemeyerCSMerleLBubakANBaxterBDGentile PoleseAColon-ReyesKet al. Olfactory and trigeminal routes of HSV-1 CNS infection with regional microglial heterogeneity. J Virol. (2024) 98:e0096824. doi: 10.1128/jvi.00968-24
FangMZhangADuYLuWWangJMinzeLJet al. TRIM18 is a critical regulator of viral myocarditis and organ inflammation. J BioMed Sci. (2022) 29:55. doi: 10.1186/s12929-022-00840-z
JiangJZhangDLiuWYangJYangFLiuJet al. Overexpression of NLRP12 enhances macrophage immune response and alleviates herpes simplex keratitis. Front Cell Infect Microbiol. (2024) 14:1416105. doi: 10.3389/fcimb.2024.1416105
EbboMCrinierAVélyFVivierE. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol. (2017) 17:665–78. doi: 10.1038/nri.2017.86
WeizmanOEAdamsNMSchusterISKrishnaCPritykinYLauCet al. ILC1 confer early host protection at initial sites of viral infection. Cell. (2017) 171:795–808.e12. doi: 10.1016/j.cell.2017.09.052
VivierEArtisDColonnaMDiefenbachADi SantoJPEberlGet al. Innate lymphoid cells: 10 years on. Cell. (2018) 174:1054–66. doi: 10.1016/j.cell.2018.07.017
HiroseSJahaniPSWangSJaggiUTormanenKYuJet al. Type 2 innate lymphoid cells induce CNS demyelination in an HSV-IL-2 mouse model of multiple sclerosis. iScience. (2020) 23:101549. doi: 10.1016/j.isci.2020.101549
WuSYFuTJiangYZShaoZM. Natural killer cells in cancer biology and therapy. Mol Cancer. (2020) 19:120. doi: 10.1186/s12943-020-01238-x
DouYXingJKongGWangGLouXXiaoXet al. Identification of the E3 ligase TRIM29 as a critical checkpoint regulator of NK cell functions. J Immunol. (2019) 203:873–80. doi: 10.4049/jimmunol.1900171
ZhangXYinZWuJXiangXZouDWangGet al. IRF4 expression by NK precursors predetermines exhaustion of NK cells during tumor metastasis. Nat Immunol. (2025) 26:1062–73. doi: 10.1038/s41590-025-02176-w
MoraruMBlackLEMuntasellAPorteroFLópez-BotetMReyburnHTet al. NK cell and ig interplay in defense against herpes simplex virus type 1: epistatic interaction of CD16A and igG1 allotypes of variable affinities modulates antibody-dependent cellular cytotoxicity and susceptibility to clinical reactivation. J Immunol. (2015) 195:1676–84. doi: 10.4049/jimmunol.1500872
DhanushkodiNRSrivastavaRPrakashSRoySCoulonPAVahedHet al. High frequency of gamma interferon-producing PLZF. J Virol. (2020) 94:e00140-20. doi: 10.1128/JVI.00140-20
TamesisRRMessmerEMRiceBADuttJEFosterCS. The role of natural killer cells in the development of herpes simplex virus type 1 induced stromal keratitis in mice. Eye (Lond). (1994) 8:298–306. doi: 10.1038/eye.1994.61
IskeleliGCamciogluYAkovaNKiranBBaharHDenizG. Lymphocyte subgroups and natural killer cell activity in recurrent herpetic stromal keratitis. Eye Contact Lens. (2008) 34:169–73. doi: 10.1097/ICL.0b013e318157a5c3
ŠutkovićJ. Neutrophils and COVID-19. Prog Mol Biol Transl Sci. (2025) 213:347–84. doi: 10.1016/bs.pmbts.2025.02.003
CaiMLiMWangKWangSLuQYanJet al. The herpes simplex virus 1-encoded envelope glycoprotein B activates NF-κB through the Toll-like receptor 2 and MyD88/TRAF6-dependent signaling pathway. PLoS One. (2013) 8:e54586. doi: 10.1371/journal.pone.0054586
Van StrijpJAVan KesselKPvan der TolMEFluitACSnippeHVerhoefJ. Phagocytosis of herpes simplex virus by human granulocytes and monocytes. Arch Virol. (1989) 104:287–98. doi: 10.1007/BF01315550
AzherTNYinXTStuartPM. Understanding the role of chemokines and cytokines in experimental models of herpes simplex keratitis. J Immunol Res. (2017) 2017:7261980. doi: 10.1155/2017/7261980
GalliSJKalesnikoffJGrimbaldestonMAPiliponskyAMWilliamsCMTsaiM. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol. (2005) 23:749–86. doi: 10.1146/annurev.immunol.21.120601.141025
MatsushimaHYamadaNMatsueHShimadaS. TLR3-, TLR7-, and TLR9-mediated production of proinflammatory cytokines and chemokines from murine connective tissue type skin-derived mast cells but not from bone marrow-derived mast cells. J Immunol. (2004) 173:531–41. doi: 10.4049/jimmunol.173.1.531
AokiRKawamuraTGoshimaFOgawaYNakaeSNakaoAet al. Mast cells play a key role in host defense against herpes simplex virus infection through TNF-α and IL-6 production. J Invest Dermatol. (2013) 133:2170–9. doi: 10.1038/jid.2013.150
DongMFitzgeraldKA. DNA-sensing pathways in health, autoinflammatory and autoimmune diseases. Nat Immunol. (2024) 25:2001–14. doi: 10.1038/s41590-024-01966-y
ChenZBehrendtRWildLSchleeMBodeC. Cytosolic nucleic acid sensing as driver of critical illness: mechanisms and advances in therapy. Signal Transduct Target Ther. (2025) 10:90. doi: 10.1038/s41392-025-02174-2
UnterholznerLKeatingSEBaranMHoranKAJensenSBSharmaSet al. IFI16 is an innate immune sensor for intracellular DNA. Nat Immunol. (2010) 11:997–1004. doi: 10.1038/ni.1932
ZhangZYuanBBaoMLuNKimTLiuYJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol. (2011) 12:959–65. doi: 10.1038/ni.2091
HornungVAblasserACharrel-DennisMBauernfeindFHorvathGCaffreyDRet al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature. (2009) 458:514–8. doi: 10.1038/nature07725
SuDHanLShiCLiYQianSFengZet al. An updated review of HSV-1 infection-associated diseases and treatment, vaccine development, and vector therapy application. Virulence. (2024) 15:2425744. doi: 10.1080/21505594.2024.2425744
LiYBieJSongCZhangTLiHZhaoLet al. SIRT2 negatively regulates the cGAS-STING pathway by deacetylating G3BP1. EMBO Rep. (2023) 24:e57500. doi: 10.15252/embr.202357500
ZhengC. Evasion of cytosolic DNA-stimulated innate immune responses by herpes simplex virus 1. J Virol. (2018) 92:e00099-17. doi: 10.1128/JVI.00099-17
ZhouCChenXPlanells-CasesRChuJWangLCaoLet al. Transfer of cGAMP into Bystander Cells via LRRC8 Volume-Regulated Anion Channels Augments STING-Mediated Interferon Responses and Anti-viral Immunity. Immunity. (2020) 52:767–781.e6. doi: 10.1016/j.immuni.2020.03.016
ZhuHZhangRYiLTangYDZhengC. UNC93B1 attenuates the cGAS-STING signaling pathway by targeting STING for autophagy-lysosome degradation. J Med Virol. (2022) 94:4490–501. doi: 10.1002/jmv.27860
YumSLiMFangYChenZJ. TBK1 recruitment to STING activates both IRF3 and NF-κB that mediate immune defense against tumors and viral infections. Proc Natl Acad Sci U.S.A. (2021) 118:e2100225118. doi: 10.1073/pnas.2100225118
JiangMChenPWangLLiWChenBLiuYet al. cGAS-STING, an important pathway in cancer immunotherapy. J Hematol Oncol. (2020) 13:81. doi: 10.1186/s13045-020-00916-z
LiuDLumKKTreenNNúñezCTYangJHowardTRet al. IFI16 phase separation via multi-phosphorylation drives innate immune signaling. Nucleic Acids Res. (2023) 51:6819–40. doi: 10.1093/nar/gkad449
KarasikAGuydoshNR. The unusual role of ribonuclease L in innate immunity. Wiley Interdiscip Rev RNA. (2024) 15:e1878. doi: 10.1002/wrna.1878
BriardBPlaceDEKannegantiTD. DNA sensing in the innate immune response. Physiol (Bethesda). (2020) 35:112–24. doi: 10.1152/physiol.00022.2019
FuYZhanXYouXNieDMaiHChenYet al. USP12 promotes antiviral responses by deubiquitinating and stabilizing IFI16. PLoS Pathog. (2023) 19:e1011480. doi: 10.1371/journal.ppat.1011480
JeffriesAMNitikaTrumanAWMarriottI. The intracellular DNA sensors cGAS and IFI16 do not mediate effective antiviral immune responses to HSV-1 in human microglial cells. J Neurovirol. (2020) 26:544–55. doi: 10.1007/s13365-020-00852-1
OmuraHOikawaDNakaneTKatoMIshiiRIshitaniRet al. Structural and Functional Analysis of DDX41: a bispecific immune receptor for DNA and cyclic dinucleotide. Sci Rep. (2016) 6:34756. doi: 10.1038/srep34756
DuanYZengJFanSLiaoYFengMWangLet al. Herpes simplex virus type 1-encoded miR-H2-3p manipulates cytosolic DNA-stimulated antiviral innate immune response by targeting DDX41. Viruses. (2019) 11:756. doi: 10.3390/v11080756
ChenHJianZXuTXuLDengLShaoLet al. Advances in the mechanism of inflammasomes activation in herpes virus infection. Front Immunol. (2024) 15:1346878. doi: 10.3389/fimmu.2024.1346878
MaruzuruYIchinoheTSatoRMiyakeKOkanoTSuzukiTet al. Herpes simplex virus 1 VP22 inhibits AIM2-dependent inflammasome activation to enable efficient viral replication. Cell Host Microbe. (2018) 23:254–265.e7. doi: 10.1016/j.chom.2017.12.014
Saïd-SadierNOjciusDM. Alarmins, inflammasomes and immunity. BioMed J. (2012) 35:437–49. doi: 10.4103/2319-4170.104408
KarabaAHFigueroaAMassaccesiGBottoSDeFilippisVRCoxAL. Herpes simplex virus type 1 inflammasome activation in proinflammatory human macrophages is dependent on NLRP3, ASC, and caspase-1. PLoS One. (2020) 15:e0229570. doi: 10.1371/journal.pone.0229570
YeFCZhouFCNithiananthamSChandranBYuXLWeinbergAet al. Kaposi’s sarcoma-associated herpesvirus induces rapid release of angiopoietin-2 from endothelial cells. J Virol. (2013) 87:6326–35. doi: 10.1128/JVI.03303-12
HatakeyamaS. TRIM family proteins: roles in autophagy, immunity, and carcinogenesis. Trends Biochem Sci. (2017) 42:297–311. doi: 10.1016/j.tibs.2017.01.002
ChabotEDurantelDLuciforaJ. TRIM proteins: A ‘swiss army knife’ of antiviral immunity. PLoS Pathog. (2025) 21:e1013147. doi: 10.1371/journal.ppat.1013147
WatanabeMHatakeyamaS. TRIM proteins and diseases. J Biochem. (2017) 161:135–44. doi: 10.1093/jb/mvw087
ZhangZBaoMLuNWengLYuanBLiuYJ. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat Immunol. (2013) 14:172–8. doi: 10.1038/ni.2492
BerubeAGmyrekGBRoyerDJCarrDJJ. Tripartite-motif 21 (TRIM21) deficiency results in a modest loss of herpes simplex virus (HSV)-1 surveillance in the trigeminal ganglia following cornea infection. Viruses. (2022) 14:589. doi: 10.3390/v14030589
SayyadZAcharyaDGackMU. TRIM proteins: key regulators of immunity to herpesvirus infection. Viruses. (2024) 16:1738. doi: 10.3390/v16111738
GuoYYGaoYZhaoYLXieCGanHChengXet al. Viral infection and spread are inhibited by the polyubiquitination and downregulation of TRPV2 channel by the interferon-stimulated gene TRIM21. Cell Rep. (2024) 43:114095. doi: 10.1016/j.celrep.2024.114095
TanTXiaL. TRIM21 aggravates herpes simplex virus epithelial keratitis by attenuating STING-IRF3-mediated type I interferon signaling. Front Microbiol. (2020) 11:703. doi: 10.3389/fmicb.2020.00703
MaXYinJQiaoLWanHLiuXZhouYet al. A programmable targeted protein-degradation platform for versatile applications in mammalian cells and mice. Mol Cell. (2024) 84:1585–1600.e7. doi: 10.1016/j.molcel.2024.02.019
XingJWengLYuanBWangZJiaLJinRet al. Identification of a role for TRIM29 in the control of innate immunity in the respiratory tract. Nat Immunol. (2016) 17:1373–80. doi: 10.1038/ni.3580
XingJZhangAZhangHWangJLiXCZengMSet al. TRIM29 promotes DNA virus infections by inhibiting innate immune response. Nat Commun. (2017) 8:945. doi: 10.1038/s41467-017-00101-w
WangJLuWZhangJDuYFangMZhangAet al. Loss of TRIM29 mitigates viral myocarditis by attenuating PERK-driven ER stress response in male mice. Nat Commun. (2024) 15:3481. doi: 10.1038/s41467-024-44745-x
WangJWangLLuWFarhatazizNGonzalezAXingJet al. TRIM29 controls enteric RNA virus-induced intestinal inflammation by targeting NLRP6 and NLRP9b signaling pathways. Mucosal Immunol. (2025) 18:135–50. doi: 10.1016/j.mucimm.2024.10.004
CossuCDi LorenzoAFiorillaITodescoAMAudritoVContiL. The role of the toll-like receptor 2 and the cGAS-STING pathways in breast cancer: friends or foes? Int J Mol Sci. (2023) 25:456. doi: 10.3390/ijms25010456
ZhangLWeiXWangZLiuPHouYXuYet al. NF-κB activation enhances STING signaling by altering microtubule-mediated STING trafficking. Cell Rep. (2023) 42:112185. doi: 10.1016/j.celrep.2023.112185
Kurt-JonesEAChanMZhouSWangJReedGBronsonRet al. Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U.S.A. (2004) 101:1315–20. doi: 10.1073/pnas.0308057100
SarangiPPKimBKurt-JonesERouseBT. Innate recognition network driving herpes simplex virus-induced corneal immunopathology: role of the toll pathway in early inflammatory events in stromal keratitis. J Virol. (2007) 81:11128–38. doi: 10.1128/JVI.01008-07
BarnettKCLiSLiangKTingJP. A 360° view of the inflammasome: Mechanisms of activation, cell death, and diseases. Cell. (2023) 186:2288–312. doi: 10.1016/j.cell.2023.04.025
CoulonPGDhanushkodiNPrakashSSrivastavaRRoySAlomariNIet al. NLRP3, NLRP12, and IFI16 inflammasomes induction and caspase-1 activation triggered by virulent HSV-1 strains are associated with severe corneal inflammatory herpetic disease. Front Immunol. (2019) 10:1631. doi: 10.3389/fimmu.2019.01631
GimenezFBhelaSDograPHarveyLVaranasiSKJaggiUet al. The inflammasome NLRP3 plays a protective role against a viral immunopathological lesion. J Leukoc Biol. (2016) 99:647–57. doi: 10.1189/jlb.3HI0715-321R
SuCZhanGZhengC. Evasion of host antiviral innate immunity by HSV-1, an update. Virol J. (2016) 13:38. doi: 10.1186/s12985-016-0495-5
XingJWangSLinRMossmanKLZhengC. Herpes simplex virus 1 tegument protein US11 downmodulates the RLR signaling pathway via direct interaction with RIG-I and MDA-5. J Virol. (2012) 86:3528–40. doi: 10.1128/jvi.06713-11
WangSWangKLinRZhengC. Herpes simplex virus 1 serine/threonine kinase US3 hyperphosphorylates IRF3 and inhibits beta interferon production. J Virol. (2013) 87:12814–27. doi: 10.1128/JVI.02355-13
WangSWangKLiJZhengC. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J Virol. (2013) 87:11851–60. doi: 10.1128/JVI.01211-13
XingJNiLWangSWangKLinRZhengC. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J Virol. (2013) 87:9788–801. doi: 10.1128/jvi.01440-13
SuCZhengC. Herpes Simplex Virus 1 Abrogates the cGAS/STING-Mediated Cytosolic DNA-Sensing Pathway via Its Virion Host Shutoff Protein, UL41. J Virol. (2017) 91:e02414-16. doi: 10.1128/JVI.02414-16
ZhangDSuCZhengC. Herpes simplex virus 1 serine protease VP24 blocks the DNA-sensing signal pathway by abrogating activation of interferon regulatory factor 3. J Virol. (2016) 90:5824–9. doi: 10.1128/JVI.00186-16
Cuchet-LourençoDAndersonGSloanEOrrAEverettRD. The viral ubiquitin ligase ICP0 is neither sufficient nor necessary for degradation of the cellular DNA sensor IFI16 during herpes simplex virus 1 infection. J Virol. (2013) 87:13422–32. doi: 10.1128/JVI.02474-13
ChristensenMHJensenSBMiettinenJJLueckeSPrabakaranTReinertLSet al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. (2016) 35:1385–99. doi: 10.15252/embj.201593458
OrvedahlAAlexanderDTallóczyZSunQWeiYZhangWet al. HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe. (2007) 1:23–35. doi: 10.1016/j.chom.2006.12.001
De PelsmaekerSRomeroNVitaleMFavoreelHW. Herpesvirus evasion of natural killer cells. J Virol. (2018) 92:e02105-17. doi: 10.1128/JVI.02105-17
LiXNayeniMMalvankar-MehtaMS. Antiviral and anti-inflammatory therapeutic interventions for treating herpes stromal keratitis: A systematic review. Ophthalmic Epidemiol. (2024) 31:191–209. doi: 10.1080/09286586.2023.2213324
BarronBAGeeLHauckWWKurinijNDawsonCRJonesDBet al. Herpetic Eye Disease Study. A controlled trial of oral acyclovir for herpes simplex stromal keratitis. Ophthalmology. (1994) 101:1871–82. doi: 10.1016/s0161-6420(13)31155-5
SchalkwijkHHShewakramaniNRDasKAndreiGSnoeckR. Combination of ganciclovir and trifluridine prevents drug-resistance emergence in HSV-1. Antimicrob Agents Chemother. (2024) 68:e0011024. doi: 10.1128/aac.00110-24
TabbaraKFAl BalushiN. Topical ganciclovir in the treatment of acute herpetic keratitis. Clin Ophthalmol. (2010) 4:905–12. doi: 10.2147/opth.s8666
KumarNSharmaSKumarRTripathiBNBaruaSLyHet al. Host-directed antiviral therapy. Clin Microbiol Rev. (2020) 33:e00168-19. doi: 10.1128/CMR.00168-19
AgelidisAMShuklaD. Cell entry mechanisms of HSV: what we have learned in recent years. Future Virol. (2015) 10:1145–54. doi: 10.2217/fvl.15.85
YamamotoMOnogiHKiiIYoshidaSIidaKSakaiHet al. CDK9 inhibitor FIT-039 prevents replication of multiple DNA viruses. J Clin Invest. (2014) 124:3479–88. doi: 10.1172/JCI73805
JaishankarDYakoubAMYadavalliTAgelidisAThakkarNHadigalSet al. An off-target effect of BX795 blocks herpes simplex virus type 1 infection of the eye. Sci Transl Med. (2018) 10:eaan5861. doi: 10.1126/scitranslmed.aan5861
HuKHarrisDLYamaguchiTvon AndrianUHHamrahP. A dual role for corneal dendritic cells in herpes simplex keratitis: local suppression of corneal damage and promotion of systemic viral dissemination. PLoS One. (2015) 10:e0137123. doi: 10.1371/journal.pone.0137123
TangRZhaiYDongLMallaTHuK. Immunization with dendritic cell-based DNA vaccine pRSC-NLDC145.gD-IL21 protects mice against herpes simplex virus keratitis. Immunotherapy. (2018) 10:189–200. doi: 10.2217/imt-2017-0060
StahlJPMaillesA. Herpes simplex virus encephalitis update. Curr Opin Infect Dis. (2019) 32:239–43. doi: 10.1097/QCO.0000000000000554
WilhelmusKRGeeLHauckWWKurinijNDawsonCRJonesDBet al. Herpetic Eye Disease Study. A controlled trial of topical corticosteroids for herpes simplex stromal keratitis. Ophthalmology. (1994) 101:1883–95. doi: 10.1016/s0161-6420(94)31087-6
RobertsCW. Comparison of diclofenac sodium and flurbiprofen for inhibition of surgically induced miosis. J Cataract Refract Surg. (1996) 22 Suppl:1, 780–7. doi: 10.1016/s0886-3350(96)80162-3
ChapellierBGuindoletDPereiraDGalettoRSahelJALabetoulleMet al. Meganuclease targeting HSV-1 protects against herpetic keratitis: Application to corneal transplants. Mol Ther Nucleic Acids. (2022) 30:511–21. doi: 10.1016/j.omtn.2022.11.006
LiYWeiYLiGHuangSXuJDingQet al. Targeting NECTIN-1 based on CRISPR/cas9 system attenuated the herpes simplex virus infection in human corneal epithelial cells in vitro. Transl Vis Sci Technol. (2022) 11:8. doi: 10.1167/tvst.11.2.8
RoyAFernandesMDasS. How much clinical practice is aligned with the Herpetic Eye Disease Study! Indian J Ophthalmol. (2021) 69:1339–40. doi: 10.4103/ijo.IJO_3210_20
VajpayeeRBDhakalBPGuptaSKSachdevMSSatpathyGHonavarSGet al. Evaluation of topical 0.03% flurbiprofen drops in the treatment of herpetic stromal keratitis. Aust N Z J Ophthalmol. (1996) 24:131–5. doi: 10.1111/j.1442-9071.1996.tb01567.x
van RooijJRijneveldWJRemeijerLVölker-DiebenHJEgginkCAGeerardsAJet al. Effect of oral acyclovir after penetrating keratoplasty for herpetic keratitis: a placebo-controlled multicenter trial. Ophthalmology. (2003) 110:1916–9. doi: 10.1016/S0161-6420(03)00798-X
Summary
Keywords
herpes simplex keratitis (HSK), herpes simplex virus type I (HSV-1), innate immunity, DNA sensor, therapeutic treatment
Citation
Nguyen P, Jacobs B, Mohanram A, Hammons C and Xing J (2025) Tug of war: innate immunity and herpes simplex keratitis. Front. Immunol. 16:1658579. doi: 10.3389/fimmu.2025.1658579
Received
02 July 2025
Accepted
15 August 2025
Published
29 August 2025
Volume
16 – 2025
Edited by
Gunnur Deniz, Istanbul University, Türkiye
Reviewed by
Umut Can Kucuksezer, Istanbul University, Türkiye
Jiaxuan Jiang, Nanjing University, China
Yun He, Nanjing Drum Tower Hospital, China
Updates

Check for updates
Copyright
© 2025 Nguyen, Jacobs, Mohanram, Hammons and Xing.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junji Xing, jxing@houstonmethodist.org
†These authors have contributed equally to this work
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.





