

The first documented function of STING was regulation of type I IFN responses, but its roles rapidly expanded to other conserved immune-related mechanisms, such as the production of pro-inflammatory cytokines or triggering non-canonical autophagy. These functions of STING are all controlled by lipid metabolism.
STING is an endoplasmic reticulum (ER)-resident protein, anchored by four transmembrane domains. It is activated downstream of cyclic GMP-AMP synthase (cGAS) upon binding its product, cyclic dinucleotides (CDNs). After activation, STING translocates to the trans-Golgi network (TGN) and perinuclear vesicles, where it activates downstream effectors (non-canonical autophagy proteins and transcription factors).
These changes in STING localization are influenced by various classes of lipids (Fig. 1). In its inactive state, sterol-binding motifs in the transmembrane and membrane proximal regions of STING facilitate ER retention through strong interaction with cholesterol. In the first step of activation, ER-based STING must bind phosphatidylinositol 3,5-bisphosphate (PtdIns(3,5)P2, also known as PIP2) alongside CDN to oligomerize1. These interactions are specifically depleted or enhanced by mechanisms that activate STING. For instance, the mammalian CDN 2’3’-cGAMP promotes the activity of sterol O-acyltransferase 1 (SOAT1)2, which leads to cholesterol clearance from the ER and a subsequent increase in STING translocation to the TGN. In addition, STING interacts with the lipid kinase complex PIKFYVE3 that produces PIP2, and STING ER-exit protein (STEEP) recruits enzymes that produce phosphatidylinositol 3-phosphate (PtdIns3P, also known as PI3P), facilitating STING translocation4. This dependency of STING activation on lipids is also hijacked by viruses to avoid innate immune activation. For example, virus-induced inflammation increases the levels of endogenous nitro-fatty acid (NO2-FA) species, which covalently modify STING by nitro-alkylation, inhibiting its palmitoylation and activation5. The precursors of NO2-FAs, polyunsaturated fatty acids (PUFAs), also bind and inhibit STING6. Similarly, 4-hydroxynonenal (4-HNE), a reactive lipid aldehyde generated during oxidative stress by lipid peroxidation, targets STING for carbonylation, preventing palmitoylation and activation7.
Fig. 1: Schematic depicting the lipid–STING crosstalk regulating STING activation, trafficking and downstream signalling.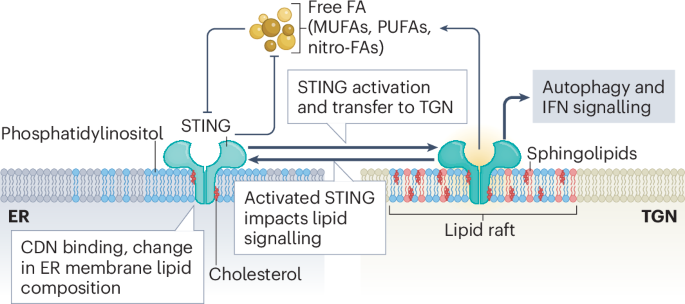
STING is embedded in the endoplasmic reticulum (ER) membrane, where pools of free fatty acids (FA), including polyunsaturated fatty acids (PUFAs), monounsaturated fatty acids (MUFAs), lipid aldehydes, and nitro-fatty acids (nitro-FAs), inhibit STING activity. In turn, STING negatively regulates the synthesis of these fatty acids. In addition to primary activating signals such as cyclic dinucleotides (CDNs), alterations in ER membrane lipid composition, especially cholesterol depletion, increase STING translocation from the ER to the trans-Golgi network (TGN). Activated STING accumulates within lipid rafts of the TGN, which are enriched in phosphatidylinositol, sphingolipids and cholesterol. In this compartment, STING initiates downstream signalling pathways, including autophagy and type I interferon (IFN) signalling. Activated STING at the TGN further promotes activation of remaining inactive STING in the ER and positively regulates lipid biogenesis and accumulation by multiple mechanisms. Lipid components of the TGN lipid rafts also modulate STING signalling activity, highlighting the bidirectional regulation between STING signalling and membrane lipid composition.
Once STING has reached the TGN, its localization and aggregation in cholesterol- and sphingolipids-enriched lipid rafts of the TGN is promoted by palmitoylation8. At this stage, the interaction with PIP2 is essential for the formation of higher-order STING oligomers and subsequent downstream signalling1. This interaction is mediated by membrane-proximal, positively charged residues in the T2-T3 linker as well as the ligand-binding domain of STING. Of note, high-order STING oligomers are better stabilized by PIP2 than PI4P (ref. 1). Whether differential mobilization of different subclasses of phosphatidylinositol adds another dimension to STING regulation by affecting its stability in different subcellular compartments needs to be investigated.
Finally, STING-dependent autophagy was also found to be regulated by myristic acid, a long-chain (LC) saturated fatty acid (SFA), which contributes to immune homeostasis by blunting cGAS–STING signalling through degradation of the STING complex9.




