




Cancer immunotherapy has revolutionized oncology by leveraging immune mechanisms to eliminate tumors. However, many cancers evade immune detection due to the absence of recognizable antigens, limiting the efficacy of current therapies.1 Redirecting pre-existing antiviral immunity toward tumors by introducing exogenous viral antigens offers a promising solution. Here, we present a novel clinically translatable approach that uses lipid nanoparticles (LNPs) to deliver mRNA encoding the measles virus hemagglutinin (H) protein directly into tumors (Fig. 1a). Prior approaches have largely relied on antibody-recruiting molecules or the delivery of defined viral peptides, thereby engaging only limited immune pathways.2 In contrast, inducing de novo expression of a full-length viral surface antigen on tumor cells enables activation of both humoral and cellular immunity through recruitment of pre-existing antibodies and presentation of multiple epitopes to CD8⁺ T cells. Compared with other viral antigens explored in mRNA–LNP-based strategies, such as the SARS-CoV-2 spike protein,3,4 measles H provides a distinct translational advantage due to the high durability and near ubiquity of measles immunity,5 supporting a robust and broadly applicable approach for redirecting antiviral immune memory toward cancer. Together, this work introduces a previously unexplored and clinically relevant framework that combines LNP-mediated mRNA delivery with a highly conserved viral antigen to broadly redirect antiviral immunity toward cancer.
Fig. 1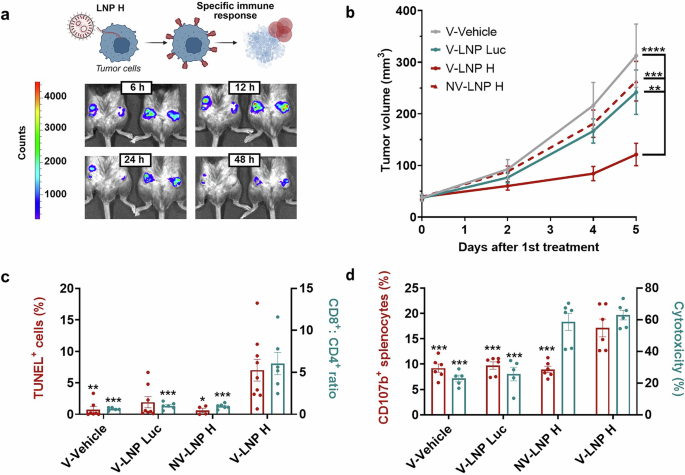 The alternative text for this image may have been generated using AI.
The alternative text for this image may have been generated using AI.
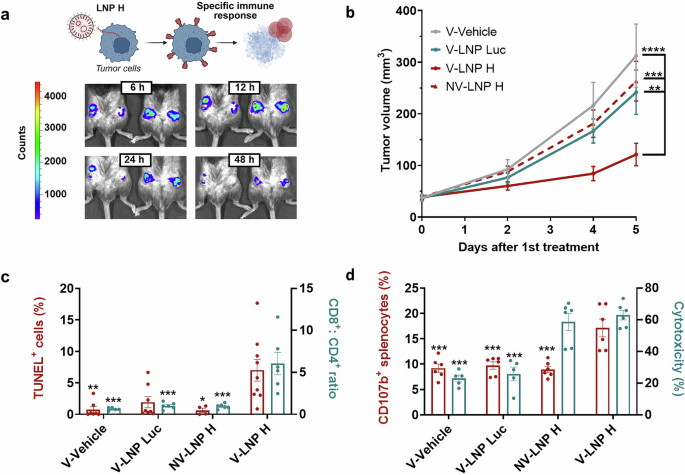
Expression of measles hemagglutinin via mRNA–LNP induces a potent CD8⁺ antitumor response in a melanoma model. a Illustration of the LNP H strategy and in vivo validation of LNP-mediated expression. Tumor cells are treated with lipid nanoparticles (LNPs) containing mRNA encoding the measles virus hemagglutinin (H) protein. Expression of H protein in cancer cells promotes immune recruitment, particularly of measles-specific CD8⁺ cytotoxic T lymphocytes, leading to tumor cell death. Bioluminescence images of B16-BL6 tumor-bearing mice at 6, 12, 24, and 48 h after intratumoral administration of LNP Luc confirm effective LNP-mediated expression at the tumor site. b LNP-mediated tumor expression of H protein drives a therapeutic response in vaccinated mice. Tumor volume progression in B16-BL6 tumors after the first intratumoral treatment (n ≥ 19). Since no therapeutic effect was observed in non-vaccinated mice regardless of treatment (NV–Vehicle, NV–LNP Luc, and NV–LNP H), we used NV–LNP H as the representative non-vaccinated control group in the current study for clarity. Tumor growth in these groups was similar to that in the vaccinated vehicle group (V–Vehicle), indicating that vaccination alone does not influence tumor growth. The vaccinated V-LNP H group exhibited significantly reduced tumor growth, whereas no effect was observed in the control groups. Statistical analysis was performed using two-way ordinary ANOVA followed by Tukey’s multiple comparison test. c LNP H treatment drives tumor cell apoptosis and a marked shift in the CD8⁺:CD4⁺ ratio. Percentage of TUNEL-positive cells in tumors (left axis, n ≥ 4 per group) and tumor-infiltrating CD8⁺:CD4⁺ ratio (right axis, n ≥ 4). Statistical analysis was performed using the Kruskal-Wallis test followed by Dunn’s multiple comparison test (TUNEL⁺ cells) and one-way ordinary ANOVA followed by Tukey’s multiple comparison test (CD8⁺:CD4⁺ ratio). Significance is shown relative to the V–LNP H group. d LNP H elicits effective antitumor activity through cytotoxic T lymphocytes. Ex vivo immune cell killing assay showing the activation of isolated splenocytes from treated mice measured via CD107b after stimulation with 16 μg/mL H protein for 5 days and co-culture with B16-BL6 cells treated with LNP H for 18 h (left axis, n = 6) and the cytotoxic activity of these splenocytes against B16-BL6 LNP H–treated cells (right axis, n = 6). Statistical analysis was performed using one-way ordinary ANOVA followed by Tukey’s multiple comparison test. Significance is shown relative to the V–LNP H group. In all subfigures, data are presented as the mean ± SEM (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). Descriptive scientific graphics were created using BioRender. V–Vehicle vaccinated mice treated with vehicle, V–LNP Luc vaccinated mice treated with LNPs containing luciferase mRNA, NV–LNP H non-vaccinated mice treated with LNPs containing H protein mRNA. V–LNP H vaccinated mice treated with LNPs containing H protein mRNA
To validate this approach, LNPs encapsulating mRNA encoding the measles virus H protein (LNP H) and, as a control, LNPs encapsulating luciferase mRNA (LNP Luc) were prepared and characterized. Both formulations exhibited an average diameter of approximately 180 nm, a low polydispersity index (PDI), a positive surface charge, and high encapsulation efficiency.
The in vitro performance of LNP H was assessed in murine (B16-BL6) and human (SK-MEL-103) melanoma models under both 2D and 3D (spheroid) culture conditions. LNP H achieved transfection efficiencies comparable to those of Lipofectamine without detectable cytotoxicity, supporting its suitability for solid tumor applications. Given widespread measles vaccination, we investigated whether SK-MEL-103 cells treated with LNPs could recruit antibodies from human serum. The majority of cells treated with LNP H successfully recruited serum antibodies, suggesting that H protein expression could initiate immune responses mediated by pre-existing antibodies in vaccinated individuals.
To further investigate the therapeutic potential in vivo, C57BL/6 mice were immunized with two doses of the measles, mumps, and rubella (MMR) vaccine to mimic human immunity. A control group received PBS instead of the vaccine. Following confirmation of successful immunization, B16-BL6 cells were subcutaneously implanted into the lower dorso-lateral region of the mice. Once tumors became palpable, vaccinated (V) animals were randomly assigned to the following groups: vehicle control (V-vehicle, baseline), LNP Luc (V-LNP Luc, carrier control), or LNP H (V-LNP H, treatment). Non-vaccinated (NV) mice were treated with LNP H (NV-LNP H) to determine whether LNP H alone, without prior measles immunity, could confer therapeutic efficacy. Each group received three doses of their respective treatment on alternate days. The in vivo transfection capability of the nanoparticles was confirmed in the V-LNP Luc group by bioluminescence emission in the tumor region (Fig. 1a). Tumor growth was monitored until the endpoint, seven days after treatment initiation. The vaccinated V-LNP H group exhibited significantly reduced tumor growth, whereas no effect was observed in the control groups (Fig. 1b). Histological analysis via TUNEL assay revealed a significant increase in apoptotic cells in tumors from vaccinated-treated (V-LNP H) animals (Fig. 1c). No systemic toxicity associated with LNP H was observed under the tested regimen, and nanoparticle activity was primarily restricted to the tumor site.
The immunological response to the treatment was characterized by analyzing tumor-infiltrating lymphocyte populations. An increase in the CD8 + :CD4+ ratio was observed in the V-LNP H group (vaccinated animals), indicating a substantial reshaping of the tumor immune landscape toward a cytotoxic T cell-dominated profile (Fig. 1c). Regulatory T cell (Treg) frequencies remained stable across groups, while natural killer (NK) cell levels were modest in all LNP-treated cohorts, indicating limited innate contribution.
The activation and cytotoxic potential of these lymphocytes were also assessed ex vivo. Splenocytes isolated from each group were stimulated with measles virus H protein for 18 hours. In the vaccinated V-LNP H group, stimulation induced a significant increase in TNF-α and IFN-γ, along with a trend toward increased granzyme B. These findings confirm that CD8⁺ T cells from V-LNP H mice showed robust recall responses to the H protein. In the non-vaccinated group, significant increases in TNF-α and a trend for IFN-γ indicate that LNP H treatment alone can partially prime the immune system; however, the absence of CD8⁺ T cell infiltration and tumor control indicates that prior vaccination is required for an effective antitumor response.
To assess cytotoxic activity, splenocytes were stimulated with measles virus H protein for 5 days and co-cultured with LNP H–treated B16-BL6 cells. Splenocytes from vaccinated treated mice (V-LNP H) displayed a significant increase in CD107b expression and cytotoxicity (Fig. 1d). This confirms that V-LNP H–primed splenocytes can effectively recognize and kill H-expressing tumor cells. Notably, splenocytes from non-vaccinated treated mice (NV-LNP H) also induced a notable reduction in cell viability, supporting partial immune priming by LNP H treatment alone.
In conclusion, this proof-of-concept demonstrates that mRNA-LNP delivery and expression of measles virus H protein can effectively redirect pre-existing antiviral immunity toward tumors, eliciting robust CD8⁺ T cell–mediated antitumor responses. The strong evidence of CD8⁺ T cell infiltration, cytokine production, and cytotoxic activity confirms that the therapeutic effect is primarily driven by cellular immunity. In addition, the recruitment of measles-specific antibodies to tumor cells expressing the H protein suggests that antibody-mediated mechanisms could contribute to tumor clearance by engaging Fc receptor–bearing effector cells. By leveraging durable and widespread immunity to a clinically validated viral antigen, this strategy offers a potentially scalable and universal approach to cancer immunotherapy. While further optimization is needed for systemic delivery and broader clinical application, the modularity of the mRNA-LNP platform provides a flexible foundation for extending this approach to other antigens and tumor types, addressing current limitations and enabling more durable, effective treatment outcomes.