



Ethics
All experimental procedures involving animals were performed in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and approved by the Animal Care and Use Committee of Peking University Health Science Center (approval no. J2024179).
Animals
Sexually mature male Wistar rats (Crl:Wl;Outbred; 200–220 g at the beginning of the experiment) and 6-week-old female Wistar rats (Crl:Wl;Outbred) were provided by the Department of Experimental Animal Sciences, Peking University Health Science Center. For the study of male fertility, we focused solely on male rats; while female rats were used only for the in vivo fertility assay. The rats were housed in separated cages with free access to standard rodent chow (SPF-F01-001, SPF Biotech, Beijing, China) and water. The room temperature was kept at 24 ± 1 °C and 40–60% humidity under a 12-h light/12-h dark cycle91. Animals are randomly allocated into treatment groups. At the end of the experiments, animals were euthanized via gradual displacement of cage air with CO2, in accordance with institutional animal welfare guidelines.
Rat model of chronic stress
Chronic forced swim (FS) is a well-established stress model that is widely employed to assess the therapeutic efficacy of antidepressant agents92. This model has been extensively utilized in neuroscience research to investigate the neurobiological mechanisms underlying depression and related disorders93,94. Prolonged exposure to the FS protocol has been demonstrated to induce cognitive impairments that closely mirror those observed in major depressive disorder (MDD)95,96. The behavioral phenotypes associated with this model, such as behavioral despair, anhedonia, and reduced motivational drive, alongside the concomitant neurochemical and endocrine alterations, are consistent with core features of depressive pathology. These changes emerge during the development of learned helplessness following repeated, inescapable stress exposure, supporting the validity of the chronic FS-stressed rat as a preclinical model of depression92,97. Moreover, chronic FS stress elicits robust neurochemical (e.g., alterations in monoaminergic, glutamatergic, and neurotrophic systems) and endocrine responses (notably activation of the hypothalamic-pituitary-adrenal axis), thereby serving as a potent psychological and physiological stressor in experimental studies98,99. In this study, chronic forced swim (FS) was used as a stressor100. Briefly, rats were placed in a glass cylinder (45 cm in height, 20 cm in diameter) filled with 30 cm depth of ice water (0–2 °C). The FS procedure was conducted to chronic FS (CFSS) group rats individually twice per day in 15-min sessions, continued for 21 consecutive days. According to the methods described elsewhere101, control rats were subjected to a sham swimming (sham FS) session by allowing them to wade in the cylinder that contained 2–4 cm of warm water (24–26 °C). Here, we used sham FS animals rather than naïve animals as control for the reason to exclude the factor of habituation for rats to the water. Rats were allowed to dry in a warm environment (30–33 °C) after swimming. The water was changed and the container was thoroughly cleaned for each rat.
Enzyme-linked immunosorbent assay (ELISA)
Blood samples were collected from the tail vein for plasma analysis of adrenocorticotropic hormone (ACTH), corticosterone (CORT), and vitamins A/E. All sampling was completed within a 2-h window (10:00 am-12:00 pm) at 24 h post-CFSS to minimize circadian interference. Plasma was separated by centrifugation (2000 × g, 20 min) after 30 min clotting at room temperature, aliquoted, and stored at −20 °C. Plasma vitamin A and E levels were detected by a rat vitamin A ELISA Kit (MERDA, Cat#M053968-48T) and rat vitamin E ELISA Kit (MERDA, Cat#M053967-48T), respectively. Rat CORT ELISA Kit (Elabscience, Cat#E-OSEL-R0002) and rat ACTH ELISA Kit (Elabscience, Cat#E-EL-R0048c) were used for plasma CORT and ACTH detection, respectively102. Hypothalamic paraventricular nucleus (PVN) tissues were homogenized in RIPA buffer with protease inhibitors. Testes were homogenized in PBS containing 0.1% butylated hydroxytoluene. After centrifugation (12,000 × g, 10 min), supernatants were analyzed using a rat CRH ELISA Kit (Elabscience, Cat#E-EL-R0270c), a rat vitamin A ELISA Kit (MERDA, Cat#M053968-48T), or a rat vitamin E ELISA Kit (MERDA, Cat#M053967-48T), with CRH and vitamin levels normalized to total protein (BCA assay, Thermo Fisher).
Open field test (OFT)
The open field test was conducted 24 h after the final chronic stress exposure. A square arena (100 × 100 × 50 cm) was placed in a dark, quiet room. Each rat was positioned at the center and allowed to freely explore for 5 min under video recording. The arena was thoroughly cleaned with 75% ethanol and dried between trials to eliminate odor cues. Behavioral parameters including total travel distance, entries into the central zone (50 × 50 cm), and time spent in the central zone were analyzed using SMART software (Panlab)103,104.
Elevated plus maze (EPM)
The elevated plus maze consisted of two open arms (50 × 10 cm), two enclosed arms (50 × 10 × 35 cm), and a central platform (10 × 10 cm) elevated 50 cm above the floor. Testing was performed 48 h post-stress. Rats were placed on the central platform facing an open arm and allowed to explore freely for 5 min. The maze was cleaned with 75% ethanol after each session. SMART software quantified total distance moved, open arm entries, and time spent in open arms103.
Sucrose preference test (SPT)
Rats were first acclimated with a 1% sucrose solution for 48 h, then subjected to a 12-hour water deprivation period before undergoing the SPT. During the test, the rats were given unrestricted access to two bottles, one containing 1% sucrose solution and the other containing tap water, for a duration of one hour. The position of the two bottles was switched midway through the test. Thereafter, consumption was measured and sucrose preference (%) was calculated. The SPT was performed both on the day prior to the CFSS treatment and 24 h after the final exposure of rats to the CFSS104.
Forced swim test (FST)
24 h after the final exposure to CFSS, rats were subjected to a 6-minute forced swimming session, during which their behavior was captured on video for subsequent analyses. The rats were considered immobile when they ceased to struggle and remained suspended in the water, making only necessary movements to keep their heads above water. The duration of immobility was recorded by an experimenter who was blinded to the experimental conditions during the last 4 minutes of the 6-minute testing session. This was done to eliminate any potential ‘first-two-minute bias’, as most animals tend to be highly active at the beginning of the FST104.
Tail suspension test (TST)
The TST was performed according to previously published methods104. In brief, a wooden box painted gray, with dimensions of 54 × 30 × 52 cm, was used as the apparatus. The rats were suspended 50 cm above the floor using adhesive tape placed ~1 cm from the tip of their tails, and the session was videotaped. Immobility was defined as complete absence of limb or body movement, except for those required for respiration, while the rat remained passively and completely still. The duration of immobility during a 6-minute test period was recorded while the rats were isolated from each other to eliminate any potential visual or acoustic associations. Observers scoring the test were blinded to the experimental conditions under which the rats were tested104.
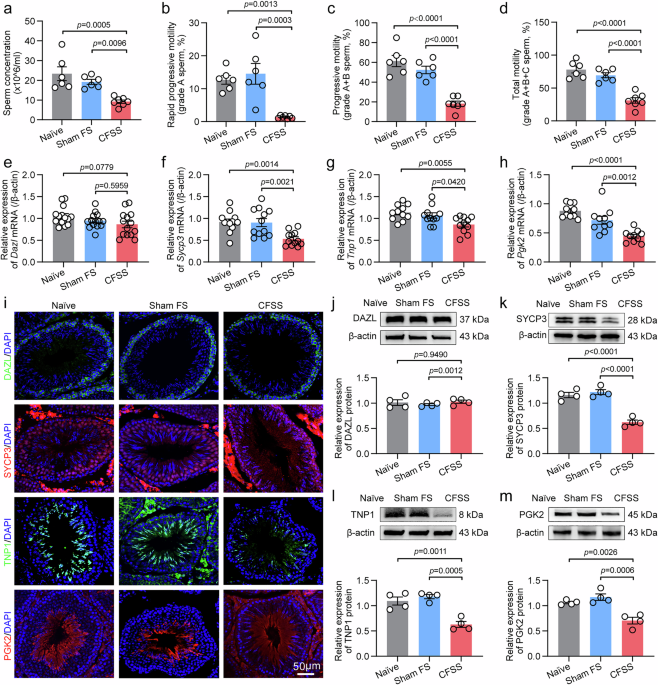
Sperm concentration, viability and motility
Cauda epididymal spermatozoa of rats were collected and prepared as previously described105. In brief, caudal epididymis were placed in high-saline (HS) solution containing 135 mM NaCl, 5 mM KCl, 2 mM CaCl2·2H2O, 1 mM MgSO4·7H2O, 20 mM HEPES, 5 mM glucose, 10 mM sodium lactate, 1 mM sodium pyruvate, adjusted to pH 7.4 with NaOH. Then, the cauda epididymis was slightly cut into three pieces, incubated at 37 °C for 10 min in a 5% CO2 incubator and the sperm was gently filtered through nylon gauze, centrifuged, and resuspended in 1 ml HS solution. A drop of the sperm suspension was used for assessment of sperm concentration and motility by CASA.
In vivo fertility assay
In vivo fertility assay was conducted following previously described methods105. Briefly, male rats were mated with 6-week-old female rats in a 1:2 ratio, where one male rat was housed overnight with two female rats. The following morning, vaginal swabs were taken from the female rats and examined under a microscope. Female rats with swabs positive for sperm were considered successfully mated and were then housed individually. After birth, the number of offspring and the pregnancy rate of these female rats were calculated and recorded.
RNA extraction and RT-qPCR
Total RNA was extracted from rat testis tissues using Trizol reagent (Life Technologies). Reverse transcription coupled with polymerase chain reaction (PCR) was executed using oligo deoxythymidine (oligo-dT) primers, in conjunction with moloney murine leukemia virus reverse transcriptase (Promega), following the manufacturer’s protocol. NanoDrop spectrophotometer was used to measure samples, recording nucleic acid concentration and A260/A280 purity ratio for rapid quantification and purity assessment. PCR primer sequences are listed in Supplementary Tables S1 and S2. Real-time quantitative PCR (RT-qPCR) assay was conducted on an ABI 7500 Fast Real-Time PCR Detection System (Applied Biosystems) employing GoTaq qPCR Master Mix (Promega). In brief, a 20 μL PCR reaction was established in strict accordance with the manufacturer’s protocol, which contains 1 μL of complementary DNA, 0.2 μM of each primer, and 10 μL of GoTaq qPCR Master Mix, with double distilled H2O (ddH2O) to adjust the final volume. Gene expression was normalized to β-actin. PCR conditions was performed with an initial 3-min incubation at 95 °C, followed by 40 thermal cycles (95 °C for 10 s, 58 °C for 20 s, and 72 °C for 10 s). The relative expression ratio of mRNA was determined using the 2−ΔΔCt method105.
Immunofluorescence staining
For the immunofluorescence staining of epididymal sperm, 20 μL of the sperm suspension was combined with an equal volume of 4% paraformaldehyde (0.1 M PB, pH 7.4). One drop of the mixture was applied to clean glass slides to create smears and then allowed to air-dry. For the immunofluorescence staining of small intestine tissues or testis tissues, rats were deeply anesthetized and underwent transcardial perfusion with 300 mL of phosphate buffer (PB, 0.1 M) followed by an equivalent volume of 4% paraformaldehyde for fixation. The removed specimens underwent post-fixation in 4% paraformaldehyde (0.1 M PB, pH 7.4) for 6 h at 4 °C, followed by cryoprotection using a 30% sucrose solution (in 0.1 M PB) at 4 °C. Several days later, 20-μm-thick sections were prepared from the tissues using a cryostat and subsequently collected onto gelatin-coated slides for immunostaining105. To perform immunostaining, small intestine, testis tissues or sperm were washed in PB (3 times × 5 min), followed by a 1-h blocking step in 10% goat or rabbit serum (0.1 M PBST with 0.3% Triton X-100) at room temperature. Intestinal tissues were incubated with rabbit anti-ZO-1 antibody (1:200, Invitrogen, Cat#40-2200, RRID: AB_2533456), mouse anti-Claudin-1 antibody (1:200, Invitrogen, Cat#37-4900, RRID: AB_2533323) or rabbit anti-Occludin antibody (1:200, Abcam, Cat#33-1500, RRID: AB_2533101) in PBST at 4 °C overnight. Testicular tissues were incubated with the primary antibody in PBST at 4 °C overnight, including rabbit anti-DAZL antibody (1:200, Abcam, Cat#ab34139, RRID: AB_731849), rabbit anti-SYCP3 antibody (1:100, Abcam, Cat#ab15093, RRID: AB_301639), rabbit anti-TNP1 antibody (1:100, Abcam, Cat#ab73135, RRID: AB_10714560), rabbit anti-PGK2 antibody (1:200, Sangon biotech, Cat#D121803, RRID: AB_3717431) or rabbit anti-STING1 antibody (1:200, Abcam, Cat#ab288157, RRID: AB_3086730). Rat spermatozoa were incubated with rabbit anti-Slc9c2 antibody (1:100, Proteintech, Cat#17398-1-AP, RRID: AB_2191353). Then, after three washes in PBS, tissues or sperm were incubated with the following appropriate secondary antibodies at room temperature for 1 h, including Alexa Fluor®488-Conjugated Donkey anti-Rabbit IgG (1:1000, Jackson Immuno Research, Cat#711-547-003, RRID: AB_2340620), Alexa Fluor®488-Conjugated Donkey anti-mouse IgG (1:1000, Jackson Immuno Research, Cat#115-545-003, RRID: AB_2338840) or Alexa Fluor®647-Conjugated Donkey anti-rabbit IgG (1:1000, Jackson Immuno Research, Cat#711-605-152, RRID: AB_2492288). The tissues or sperm were counterstained with the nuclear marker DAPI (100 ng/mL) carrying blue fluorescence for 10 min at room temperature. The slides were mounted in Gel-Mount medium. Visualization of fluorescence signal was performed by Leica Stellaris 5 microscopy (Leica Microsystems, Wetzlar, Germany) at excitation wavelengths of 488 nm (green), 647 nm (red) or 405 nm (blue), respectively. At least three fields per slide were analyzed to establish reproducibility105.
Western blot
Rat testicular tissues, small intestine tissues or sperm cells were homogenized in ice-cold lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM PMSF). Homogenates were centrifuged (12,000 × g, 10 min, 4 °C), and supernatants were analyzed with BCA assay (Pierce). Equal protein amounts (40 μg) were separated on 10% SDS-PAGE gels, transferred to PVDF membranes (Bio-Rad), and blocked with 5% nonfat milk/BSA in TBST. Membranes were incubated overnight at 4 °C with primary antibodies, including rabbit anti-DAZL antibody (1:1000, Abcam, Cat#ab34139, RRID: AB_731849), rabbit anti-SYCP3 antibody (1:1000, Abcam, Cat#ab15093, RRID: AB_301639), rabbit anti-TNP1 antibody (1:500, Abcam, Cat#ab73135, RRID: AB_10714560), rabbit anti-PGK2 antibody (1:2000, Sangon biotech, Cat#D121803, RRID: AB_3717431), rabbit anti-STING1 antibody (1:1000, Abcam, Cat#ab288157, RRID: AB_3086730), or mouse anti-β-actin antibody (1:2000, Immunoway, Cat#YM3028, RRID: AB_2629465) for rat testis tissues. Rabbit anti-ZO-1 antibody (1:1000, Invitrogen, Cat#40-2200, RRID: AB_2533456), mouse anti-Claudin-1 antibody (1:1000, Invitrogen, Cat#37-4900, RRID: AB_2533323), rabbit anti-Occludin antibody (1:1000, Abcam, Cat#33-1500, RRID: AB_2533101), or mouse anti-β-actin antibody (1:2000, Immunoway, Cat#YM3028, RRID: AB_2629465) for rat small intestine tissues. Rabbit anti-Slc9c2 antibody (1:500, Proteintech, Cat#17398-1-AP, RRID: AB_2191353) and mouse anti-β-actin antibody (1:2000, Immunoway, Cat#YM3028, RRID: AB_2629465) for rat spermatozoa. The blots were washed in TBST and then were incubated in horseradish peroxidase-conjugated secondary antibody, including goat anti-rabbit IgG-HRP (1:2000, Santa Cruz, Cat# sc-2004, RRID: AB_631746) or goat anti-mouse IgG-HRP (1:2000, Santa Cruz, Cat# sc-2005, RRID: AB_631736). Protein bands were visualized using an enhanced chemiluminescence detection kit (Pierce) followed by autoradiography using Hyperfilm MP (Santa Cruz). The bands, including all the target protein and the internal loading control (β-actin), were quantified with a computer-assisted imaging analysis system (Image J, NIH), and the normalized ratio of the target protein to β-actin band density was used to calculate the alteration of corresponding protein expression105.
Exogenous CORT and RU-486 administration
CORT (Twbio, Cat#DA88478) was dissolved in absolute ethanol and diluted with saline (1:19 ratio) to a final concentration of 2 mg/kg106. The vehicle control comprised 5% ethanol in saline. RU-486 (KIRGEN, Cat#IM320) was suspended in 5% DMSO, 5% Cremophor EL, and 90% ddH2O, with its vehicle containing the same solvent mixture38. For exogenous CORT administration in normal rats, adult male Wistar rats were randomly divided into four groups as follows: vehicle group received 1 mL of CORT vehicle (5% ethanol in saline) daily; CORT group was administered 2 mg/kg CORT solution daily; CORT + RU-486 group was co-treated with 2 mg/kg CORT and 3 mg/kg RU-486 suspension daily; CORT + RU-vehicle group received 2 mg/kg CORT plus RU-486 vehicle daily. For exogenous RU-486 administration to CFSS model rats, adult male Wistar rats undergoing CFSS were divided into three groups as follows: CFSS+vehicle received RU-486 vehicle (5% DMSO, 5% Cremophor EL, 90% ddH2O) daily; CFSS + RU-486 was administered 2 mg/kg RU-486 suspension via oral gavage daily; CFSS + RU-vehicle was treated with RU-486 vehicle daily. All treatments were delivered via oral gavage once daily for 21 consecutive days.
ABX cocktail treatment and FMT
Male Wistar rats (recipient rats) were given a sterile-water drink containing: 1 g/L ampicillin (Songon, Cat#A610028), 1 g/L neomycin (Songon, Cat#A610366), 1 g/L metronidazole (Songon, A600633), and vancomycin 0.5 g/L (Songon, Cat#A600983) for seven days starting from the initiation of the experiment. Fresh fecal pellets (200 mg/day) from sham FS and CFSS donor rats were homogenized in sterile saline (1:10 w/v), allowed to sediment for 15 min at room temperature, and filtered through sterile gauze. Sham-FMT and CFSS-FMT recipient rats received 1 mL of respective donor fecal suspensions via oral gavage once per day for 21 days91.
Lactobacillus supplementation
For Lactobacillus supplement to CFSS group rats, Lactobacillus (BNCC, Cat#223759), which composition is Lactobacillus sp. enrichment culture clone MP30 16S ribosomal RNA gene, were cultured in MRS medium supplemented with 5% FPS under anaerobic conditions at 37%. Perform serial dilutions of the bacterial culture, spread onto agar plates, incubate, then count colonies on plates containing 30-300 colonies, and calculate the number of viable bacteria per milliliter (CFU/mL) based on the dilution factor. CFSS group rats were colonized with 200 μL indicated bacteria consortia at a dose of 2 × 108 colony-forming units per 200 μL suspended in the MRS medium or the same volume of MRS medium by oral gavage once per day for 21 days107.
16S rDNA amplicon sequencing and bioinformatics analysis
Small intestine digesta samples were immediately snap-frozen and stored at −80 °C. Bacterial DNA was extracted using the MagPure Soil DNA LQ Kit (Magen, China), with concentration and integrity assessed by NanoDrop 2000 spectrophotometry and agarose gel electrophoresis. The V3-V4 hypervariable regions of the 16S rRNA gene were amplified via PCR (25 μL reaction volume) using universal primers 343 F (5’-TACGGRAGGCAGCAG-3’) and 798 R (5’-AGGGTATCTAATCCT-3’), incorporating Illumina adapters and sample-specific barcodes. Amplified fragments were purified with Agencourt AMPure XP beads (Beckman Coulter), quantified via Qubit dsDNA assay, and sequenced on an Illumina NovaSeq6000 platform (250 bp paired-end reads; OE Biotech Co., China). Raw reads were preprocessed with Trimmomatic to remove low-quality sequences (Q20 threshold) and assembled using FLASH (10–200 bp overlap; ≤20% mismatch). Chimeric sequences were filtered by VSEARCH, followed by clustering into operational taxonomic units (OTUs) at 97% similarity via QIIME (v1.8.0). Representative OTU sequences were taxonomically annotated against the SILVA database (v132) using the RDP classifier (70% confidence). Alpha diversity (Chao1 and Shannon indices) and beta diversity (unweighted UniFrac PCoA) analyses were performed in QIIME, with phylogenetic trees generated from UniFrac distance matrices91. Sequencing and bioinformatics were conducted by OE Biotech Co., Ltd. (Shanghai, China).
Metabolomics profiling and bioinformatics analysis
Small intestine digesta (n = 10 biological replicates per group), plasma (n = 10 biological replicates per group), and testis (n = 4 biological replicates per group) samples stored at −80 °C were thawed on ice. Plasma was separated by centrifugation (2000 × g, 20 min) after 30 min clotting at room temperature. For solid samples, 20 mg of material was homogenized in 400 μL methanol:water (7:3, v/v) containing internal standards, vortexed, sonicated (ice bath, 10 min), and centrifuged (12000 × g, 4 °C). For liquid samples, 50 μL was mixed with 300 μL ACN:methanol (1:4, v/v) with internal standards, followed by vortexing and centrifugation. Supernatants from both sample types were filtered (0.22 μm) and analyzed via LC-MS using a Shimadzu LC20 UPLC system coupled with an AB Sciex Triple TOF-6600 mass spectrometer (Waters ACQUITY UPLC HSS T3 C18 column, 1.8 μm, 2.1 × 100 mm; 40 °C; 0.4 ml/min flow rate). The mobile phase consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B), with a gradient from 5% to 90% B over 11 min, held for 1 minute, and then come back to 5% mobile phase B within 0.1 minute, held for 1.9 minutes. Key settings included: IonSpray Voltage, 5500 V (ESI + )/ -4500 V (ESI-); Ion Source Gas 1 and 2, 50/60 psi; Curtain Gas, 35 psi; Source Temperature, 550 °C; and Collision Energy, 30 eV/ -30 eV. Raw data (mzML format, converted by ProteoWizard v3.0) underwent peak extraction and alignment using XCMS, with SVR-based area correction and removal of low-frequency peaks ( < 50% detection rate). Metabolites were identified against in-house and public databases (SILVA, AI, MetDNA). For bioinformatics, PCA and hierarchical clustering (HCA, heatmaps with dendrograms) were performed in R v3.5.1 (unit variance scaling). Differential metabolites were defined by VIP > 1 (OPLS-DA, MetaboAnalystR v1.0.1) and P < 0.05 (Student’s t-test), validated via permutation testing (200 cycles). KEGG pathway enrichment was assessed using a hypergeometric test on annotated metabolites91. Representative total ion chromatograms (TICs) or Base Peak Chromatogram (BPC) for the methods are provided in Supplementary Fig. S23.
Vitamin A and Vitamin E supplementation
The procedure of vitamin A and vitamin E supplementation was performed as described in previous studies108,109. In brief, vitamin A palmitate and vitamin E acetate were dissolved in corn oil, supplemented with 0.3% Tween 80 (v/v) and 0.5% sodium carboxymethyl cellulose (CMC-Na, w/v). For vitamin A/E supplement, CFSS model rats received daily oral gavage of 1 mL emulsion containing corn oil, ddH2O, 0.3% Tween 80, and 0.5% CMC-Na (without vitamins) for 21 consecutive days, while the vitamin A/E supplementation group was administered 1 mL emulsion containing the same solvent components plus 2000 IU/kg vitamin A palmitate108 and 50 mg/kg vitamin E acetate109 for the same duration. Here, the duration of 21-days treatment was used for the reason that the total duration of the spermatogenesis is 50 days for rats, and 3 weeks treatment was almost covered half of one spermatogenesis duration110.
Glycoursodeoxycholic acid (GUDCA) supplementation
For GUDCA intervention, CFSS rats in the treatment group received daily oral gavage of 1 mL phosphate-buffered saline (PBS) containing 60 mg/kg glycoursodeoxycholic acid (GUDCA, Acmec, T91840)107 for 21 consecutive days. In parallel, CFSS rats in the control group received daily oral gavage of 1 mL PBS alone over the same period.
Comprehensive supplementation of Lactobacillus and vitamin A/E
CFSS rats in the comprehensive therapy group received daily oral gavage of 200 μL containing a defined bacterial consortium (Lactobacillus), along with 1 mL of vitamin A palmitate (2000 IU/kg) and vitamin E acetate (50 mg/kg) for 21 consecutive days. In contrast, CFSS rats in the control group received daily gavage of 200 μL MRS medium and 1 mL vitamin-free emulsion over the same period.
RNA sequencing and data analysis
Total RNA was extracted from testis or epididymis tissues of male rats after CFSS treatment. The Beijing Genomics Institute (BGI) conducted the RNA sequencing (RNA-seq) for this study. In briefly, total RNA was extracted using TRIzol reagent as per the manufacturer’s instructions following the dissociation of testis or epididymis tissues. Isolation of total RNA was achieved using the RNeasy mini kit (Qiagen, Germany). Subsequently, TruSeq RNA Sample Preparation Kit (Illumina, USA) was employed to prepare the RNA-seq library from 1 µg of total RNA. Briefly, following the purification of poly A-containing mRNA, the molecules were cleaved into small fragments via divalent cations at 94 °C for 8 min. Subsequently, library concentration was determined with a Qubit 2.0 Fluorometer (Invitrogen, USA) and verified via an Agilent 2100 bioanalyzer (Agilent Technologies, USA) to assess the insert size and to establish the mole concentration. To identify differentially expressed genes (DEGs) across groups, gene expression levels were normalized to TPM (Transcript per kilobase per million mapped reads). For inter-group comparisons, the binary logarithm of the fold change (log2(FC)) was calculated based on these TPM values. Significant differences in gene expression were defined using a log2(FC) cutoff of ≥ 1.0, which corresponds to a minimum of two-fold change. Following transcriptome assembly, we determined the expression levels for all identified genes and transcripts. Functional enrichment was ultimately conducted using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) databases to elucidate the biological roles of the DEGs. GO analysis categorized these genes into biological processes, molecular functions, and cellular components. Simultaneously, KEGG pathway analysis identified critical signal transduction and disease-related mechanisms, providing a comprehensive functional framework for the identified genes and their associated pathways91.
Plasmid construction and lentivirus infection
Lentivirus-expressing Sting1 (LV-Sting1) and lentivirus-expressing shRNA targeting rat Sting1 (LV-shSting1), both linked with an EGFP tag, using either the pcSLenti-EF1-EGFP-CMV-MCS-WPRE or pCLenti-U6-shRNA-CMV-EGFP-WPRE vector were produced by OBiO technology (Shanghai, China). For Sting1 silencing, three shRNA sequences were designed (Supplementary Data 1), with preliminary experiments demonstrating that shRNA (Sting1)-1 effectively suppressed Sting1 expression in testis tissues, leading to its selection for further study. Lentivirus-expressing Slc9c2 (LV-Slc9c2) and lentivirus-expressing shRNA targeting rat Slc9c2 (LV-shSlc9c2) fused to an EGFP tag, using pcSLenti-EF1-EGFP-CMV-MCS-WPRE and pCLenti-U6-shRNA-CMV-EGFP-WPRE vector, were provided by BrainVTA technology (Wuhan, China). Lentiviral titer was determined by qPCR quantification of viral DNA copies integrated into the host genome. Genomic DNA was extracted from 293 T cells (ATCC, Cat# CRL-3216) 72 h post-infection, and qPCR was performed targeting a viral-specific sequence (e.g., WPRE). The copy number was calculated using a standard curve and converted to transducing units per milliliter (TU/mL) based on the number of infected cells, volume of virus used, and gDNA parameters. Three shRNA sequences for Slc9c2 were evaluated (Supplementary Data 1), and shRNA (Slc9c2)-3 was selected based on its superior silencing efficacy in preliminary experiments. For testis Sting1 overexpression or spermatozoa Slc9c2 knockdown in naïve rats, LV-Sting1 lentivirus or LV-shSlc9c2 lentivirus were injected through the efferent ductule of testis at a final titer of 5 × 108 transducing units/ml. The procedure of intratesticular virus injections were performed as described elwhere with minor modification111,112,113. Briefly, rats were anesthetized via intraperitoneal injection of pentobarbital sodium (50 mg/kg). The surgical site was prepared aseptically using sequential disinfection with 70% ethanol and povidone-iodine solution. A midline ventral incision, ~1.5 cm anterior to the genital apparatus, was made through the skin and abdominal wall using sterile surgical scissors under aseptic conditions. The testes were exteriorized by gentle traction on the epididymal fat pad. Each testis was stabilized with fine forceps, and lentiviral particles were delivered directly into the seminiferous tubules via the efferent ducts using a 33-gauge microsyringe (Hamilton, Switzerland). A volume of 50 μL containing 0.04% Trypan blue, used as a tracer to visualize distribution, was injected into the interstitial compartment of both left and right testes, to ensure consistent delivery volume and location within the testis or efferent ductule. Following successful delivery, the abdominal wall and skin incisions were sutured in layers using sutures, respectively111,112. 21 days after viral infection, a transfection efficiency of lentivirus was routinely achieved as observed under fluorescence microscopy. For testis Sting1 knockdown or spermatozoa Slc9c2 overexpression of CFSS-treated rats, LV-shSting1 lentivirus or LV-Slc9c2 lentivirus was injected through the efferent ductule of testis at a final titer of 5 × 108 transducing units/ml (total volume of 50 μL in both left and right side of the testis tissues). The CFSS procedure was initiated 2–3 days after lentiviral injection to allow initial recovery from surgical trauma while avoiding interference with viral transduction.
Statistical analysis
The sample size was determined based on previously described guidelines114,115. In brief, the total number of animals was selected according to the number of experimental groups and the specific requirements of each assay. For instance, n = 6–8 rats per group were used for qPCR and RNA-seq analyses, whereas n = 3–6 rats per group were sufficient for Western blot experiments, in accordance with established standards for statistical power and experimental reproducibility. Statistical analyses were performed with GraphPad Prism 9.0 for Windows (GraphPad Software, La Jolla, CA). The number of independent biological replicates are provided in the figures and figure legends. All quantitative biochemical data and immunofluorescence staining are representative of at least three independent biological replicates. Shapiro–Wilk tests were used to assess normality in the distribution (Gaussian distribution, p > 0.05, Levene’s post hoc test p > 0.05) for each group, and only the data were normally distributed and variances were similar between groups to be compared were subjected to parametric statistical tests. Two-tailed unpaired Student’s t test was used for the comparison of the mean values between two groups. One-way analysis of variance (ANOVA) with Tukey’s post hoc test was used for multiple comparison. All data are reported as means ± SEM, and differences with p < 0.05 were considered statistically significant. The investigators were blinded to group allocation during data collection. All statistical data are presented in Supplementary Data 2.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.